J Biomed 2017; 2:64-77. doi:10.7150/jbm.18877 This volume Cite
Review
Nanoparticle Design Strategies for Effective Cancer Immunotherapy
Praveena Velpurisiva1, Aniket Gad1, Brandon Piel2, Rahul Jadia1, Prakash Rai2 ![]()
1. Department of Biomedical Engineering and Biotechnology, University of Massachusetts Lowell, USA.
2. Department of Chemical Engineering, University of Massachusetts Lowell, USA.
Received 2016-12-22; Accepted 2017-2-1; Published 2017-2-25
Abstract

Cancer immunotherapy is a rapidly evolving and paradigm shifting treatment modality that adds a strong tool to the collective cancer treatment arsenal. It can be effective even for late stage diagnoses and has already received clinical approval. Tumors are known to not only avoid immune surveillance but also exploit the immune system to continue local tumor growth and metastasis. Because of this, most immunotherapies, particularly those directed against solid cancers, have thus far only benefited a small minority of patients. Early clinical substantiation lends weight to the claim that cancer immunotherapies, which are adaptive and enduring treatment methods, generate much more sustained and robust anticancer effects when they are effectively formulated in nanoparticles or scaffolds than when they are administered as free drugs. Engineering cancer immunotherapies using nanomaterials is, therefore, a very promising area worthy of further consideration and investigation. This review focuses on the recent advances in cancer immunoengineering using nanoparticles for enhancing the therapeutic efficacy of a diverse range of immunotherapies. The delivery of immunostimulatory agents to antitumor immune cells, such as dendritic or antigen presenting cells, may be a far more efficient tactic to eradicate tumors than delivery of conventional chemotherapeutic and cytotoxic drugs to cancer cells. In addition to its immense therapeutic potential, immunoengineering using nanoparticles also provides a valuable tool for unearthing and understanding the basics of tumor biology. Recent research using nanoparticles for cancer immunotherapy has demonstrated the advantage of physicochemical manipulation in improving the delivery of immunostimulatory agents. In vivo studies have tested a range of particle sizes, mostly less than 300 nm, and particles with both positive and negative zeta potentials for various applications. Material composition and surface modifications have been shown to contribute significantly in selective targeting, efficient delivery and active stimulation of immune system targets. Thus, these investigations, including a wide array of nanoparticles for cancer immunotherapy, substantiate the employment of nanocarriers for efficacious cancer immunotherapies.
Keywords: nanomedicine, monoclonal antibody, cancer vaccine, drug delivery, immunology, immunosuppression, immunostimulation, immunomodulation, immunotheranostics, virus-based vaccine
Introduction
With the recent launch of the Cancer MoonShot 2020 program, the development of better and targeted treatments continues in our long and drawn out war against cancer. The most commonly used treatments in cancer are surgery, radiation therapy, chemotherapy or some combination of the three, all of which have shown significant improvements as cancer treatments. Despite these advancements, surgery, when possible, continues to offer the best results. However, cancer is usually detected at late stages when the disease has already spread, limiting surgery-based treatments mostly to early cancers. Chemotherapy and radiation therapy often fall short and have higher cancer relapse rates [1]. This has encouraged many researchers to focus on employing the human body's own defense system as a tool to combat cancer. Cancer immunotherapy has shown promise as an alternative or an extension to the current forms of cancer treatment, particularly in fighting against malignant tumors [2, 3].
The immune system plays a significant role in identifying incipient neoplasia leading to tumorigenesis, and in eliminating certain types of tumors [4, 5]. Despite that, cancer cells have the ability to evade immune response, and often, immune cells are exploited for tumor growth and progression [6]. However, cancer immunotherapy focuses on recruiting tumor killing immune cells already present in the tumor microenvironment, initiating an immune response. Both innate and the adaptive immunity can be primed to identify and expunge cancer cells.
There are currently two main ways to initiate immunotherapy. The first is to inject the patient with a tumor-specific antibody or, more often, an antigen to train the host immune system to identify cancerous cells. The other is to grow cells ex vivo that are able to target and destroy cancer cells. In a recent clinical case study, Tran et al. grew CD8+ T cells which target a mutant form of KRAS (KRAS G12D), a common “hotspot” mutation among colorectal cancers. A patient with seven KRAS G12D positive metastasized lung tumors was injected with these T cells. After two weeks, all tumors had regressed and the patient was discharged. After 90 days, only two of the tumors relapsed. These tumors were removed surgically. Sequencing of the relapsed tumors showed that resistance to the treatment occurred due to a deletion of chromosome 6, on which the mutant KRAS gene resides [7].
Immunotherapy can also be used in combination with other cancer therapies such as chemotherapy, phototherapy, and gene therapy [3, 8-12]. The potential of cancer immunotherapy was officially recognized by the Food and Drug Administration (FDA) when it approved Sipuleucel-T (Provenge®) for castration-resistant, metastatic prostate cancer. Additional FDA approvals for cancer immunotherapy are given below in table 1. Several antibodies have also received approval for treatment of previously listed types of cancer [13]. One such antibody, Ipilimumab, has shown promising clinical trials; it binds to CTLA-4 antigens present in tumor infiltrating regulatory T cells (T-reg cells), thereby enhancing antitumor activity [14]. However, the issue with the current immunotherapies is that they are unaffordable. For example, the Provenge vaccine treatment costs $93,000 whereas an Ipilimumab treatment cycle amounts to about $120,000 [15-18]. This raises the price tags in an already expensive health care system.
A list of cancer immunotherapies that are either already approved or in clinical trials is shown in table 1.
List of cancer immunotherapy drugs approved or in clinical trials
| Drug / Treatment | Type of Cancer | Year | Approval |
|---|---|---|---|
| Nivolumab [19] | Relapsed Lymphoma | 2016 | Approved |
| Keytruda [20] (Pembrolizumab) | Lung Cancer | 2015 | Approved |
| Blincyto [21] (blinatumomab) | Leukemia | 2014 | Approved |
| Ipilimumab [22] | Melanoma | 2011 | Approved |
| Provenge [23] | Prostate Cancer | 2010 | Approved |
| Interleukin 2 [24] | Renal Cell Carcinoma Melanoma | 1992 1998 | Approved |
| R-IDARAM [26] | Central Nervous System Lymphoma | NA | Phase III |
| Avelumab [27] | Lung Cancer | NA | Phase III |
| Bruton's Tyrosine Kinase (BTK) inhibition [28] | B-cell Lymphoma | NA | Phase I |
*NA = Not Approved
Several measures are explored to reduce the cost of the immunotherapy treatment. Integrating nanotechnology for cancer immunotherapy can substantially reduce the cost, increase the therapeutic efficacy of the delivery, and reduce localized systemic toxicity [29]. However, it is important to take into consideration several critical parameters before selecting the drug delivery system (DDS) for cancer immunotherapy. The type and nature of the DDS needs to be investigated prior to selecting the type of cancer immunotherapy. There have been reports showing that certain types of nanoparticles such as carbon nanotubes, quantum dots, and superparamagnetic iron oxide, can themselves be immunogenic, induce toxicity, and/or cause cancer in certain animal models [30-32]. The most commonly used DDSs utilize biodegradable lipid or polymer based nanoparticles [33]. Many studies have also focused on using gold and silica nanoparticles for cancer immunotherapy [34, 35].
The physicochemical characteristics of the nanoparticles, such as size, shape, surface charge, and surface functionalization, are crucial parameters in designing DDS for cancer immunotherapy [36]. The delivery mechanism of nanoparticles depends greatly on enhanced permeation and retention effects. Nanoparticles less than 100 nm show less restriction to tumor penetration but it is preferred to use nanoparticles smaller than 50 nm for elevated therapeutic effects [37]. Some studies on nanoparticle shape showed that elongated nanoparticles have greater internalization potential compared to their spherical or cuboidal counterparts [38, 39]. Surface charge is also an important parameter in designing the DDS, predominantly for in vivo studies, to avoid any systemic toxicity. Surface functionalization offers even more specificity by actively targeting the nanoparticles to the desired site. Surface functionalization also enables binding of PEG (poly-ethylene-glycol), which increases the circulation time. The variability offered by drug delivery systems can be used to deliver antigens or adjuvants in order to induce various types of immune responses [34]. The following schematic presents the approach using nanoparticles for cancer immunotherapy (Figure 1):
In cancer vaccine type immunotherapy, nanoparticles can be used to deliver tumor-specific antigens to activate antigen-presenting cells (APCs). Once activated, the APCs can further activate effector cells, thereby inducing an antigen-specific immune response against the cancer cells. In a different immunotherapy, selective targeting can be achieved by the conjugation of monoclonal antibodies (mAbs) to the surface of nanoparticles, enabling the nanoparticle to directly bind to the cancer cells. Certain antibodies can also be used to suppress the function of T-reg cells. T-reg cells inhibit the function of immune killer cells, or mature the dendritic cells (DCs), to induce cell death via cytotoxic T lymphocyte (CTL) recruitment [40]. For non-specific immunotherapy, the enhanced delivery of certain adjuvants, such as cytokines, can be achieved with the help of nanoparticles to boost the immune response in conjunction with other cancer immunotherapies. Nanoparticles also benefit from theranostic applications including delivery of imaging agents along with the therapeutic moieties to diagnose and track the treatment [41, 42].
Among the different types of immunotherapies specified, the following therapies detail the recent in vivo research. A few are currently undergoing clinical trials and some have been approved by the FDA.
DC-based Immunotherapy
A majority of recent nano-immunotherapy research has been dedicated towards the targeting of DCs. As the main APCs, they play a prominent role in early initiation of immune responses by CTL-activation [43, 44, 47, 48]. Although DCs are present abundantly in the lymph nodes, their antigen presentation property is often affected by low cellular uptake of antigens [45, 48] or low lymphatic drainage of micro-sized moieties [60], thereby altering CTL activation [45, 48]. Implementation of nanoparticles and exploiting their physicochemical properties for delivery of antigens and adjuvants has shown promising results in DC-based immunotherapy by elevating CTL responses [47].
Role of nanoparticles in cancer immunotherapy (Arrows indicate pathway to immune response).
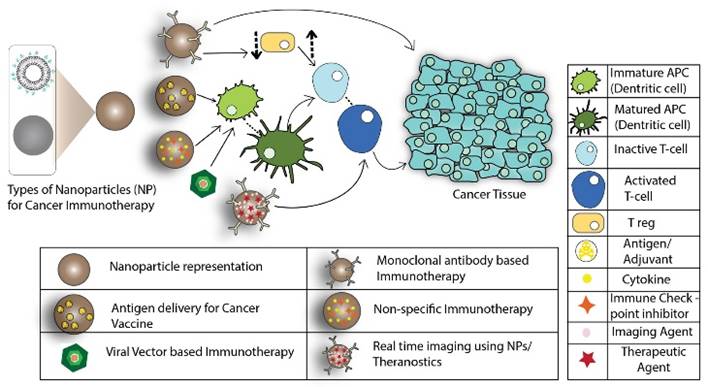
Summary of key design characteristics of the different types of nanoparticles used in cancer immunotherapy.
| Ref # | Nanoparticle Type | Size (nm) | Surface Charge (mV) | Surface Modification | Tumor Model/ Cell Line | Type of Immunotherapy |
|---|---|---|---|---|---|---|
| [43] | Lipoplex | 200 to 320 | 0 to -30 | None | CT26 colon carcinoma B16 lung model | Dendritic Cell- based Immunotherapy |
| [44] | Poly (lactic-co-glycolic acid) | 42.3 ± 25 | -38 ± 1.7 | CD40-mAb | B16-OVA melanoma mouse model | |
| [45] | Liposome | 30 | * | ɑ-Ap antigen peptide | EG.7-OVA | |
| [46] | Upconversion Nanoparticles | 170 | 10 | PAA-PEG-PEI-OVA | C57BL/6 mouse | |
| [47] | Poly (propylene sulfide) | 30 | * | OVA/ TRP-2/ TLR9 ligand CpG DNA | B16-F10 melanoma model E.G7-OVA model mouse, lymphoma cell line | |
| [48] | Poly (propylene sulfide) | 30 | * | Pyridyl disulfide-NPs/ CpG conjugation | B16-F10 melanoma mouse model | |
| [49] | Poly (lactic-co-glycolic acid) | 110 | -35 | Cancer cell membrane (MPLA) | B16-F10 mouse melanoma model | |
| [50] | Micelles | 26.7± 6 | Positive | Thiolated -OVA/CpG oligodeoxynucleotides (ODNs) | C57BL/6 mice, mouse dendritic cell line DC2.4 | |
| [51] | Lipid/calcium/ phosphate | 45 | 15 | Mannose-PEG | B16-F10 melanoma metastatic model | |
| [41] | poly (isobutylene-alt-maleic anhydride) (PIMA) + Fluorescence Resonance Energy Transfer (FRET) pair | 186.8 ± 7.3 | -41.5 | Anti-PD-L1 | B16-F10 melanoma model, 4T1 breast cancer model, BALB/c mice with DU15 and DU15TR prostate cancer model | Chemo-Immunotherapy |
| [35] | Porous silicon | 171 | 11.6 ± 1.1 | anti-CD326 Ab | MCF-7, MDA-MB-231 | |
| [52] | Poly (propylene-imine) dendrimers | 4.5 G** | * | mAbK1 | Ovarian cancer model | |
| [53] | Cationic polyplex | * | Positive | Attenuated Salmonellae | C57BL/6-derived B16 melanoma mouse model | Oral DNA vaccine |
| [54] | Empty Cowpea mosaic virus (eCPMV) | 30 | * | None | B16F10 metastatic lung model. Mice with 4T1 breast tumors. Mice with CT26 colon tumor. Mice with ovarian carcinoma. | Viral Immunotherapy |
| [55] | Gold | 8.5 | -30.14 | SM5-1 mAb | Hepatocellular carcinoma: HCC-LM3-fLuc cells in BALB/c mice. | Membrane protein, p230 targeting using mAbs |
| [56] | PEGylated Lipid nanocapsules | 65 ± 2 | -3 ± 1 | None | The EG7-OVA lymphoma and B16-F10 melanoma mouse models | Myeloid Derived Suppressor Cell-targeting |
| [57] | Gold | 5, 30 and 80 | * | Thiolated- CpG oligodeoxynucleotides (ODNs) | B16-OVA tumor mouse model | Macrophage Targeting |
| [58] | Cobalt oxide nanoparticles | 72 ± 8 | -37 | Phosphonomethyl iminodiacetic acid | Swiss mice tumor- free | |
| [59] | Bovine Serum Albumin | 34, 153 and 224 | * | None | Mouse melanoma model | Neutrophil Targeting |
| [60] | Iron-oxide particles | 50 to 100 | * | Dextran | B16 melanoma mouse model | CTL Activation |
| [60] | Quantum dot nanocrystals | 30 | * | Avidin | B16 melanoma mouse model | |
| [61] | Hydroxy-poly(ethylene glycol) | 246.8 ± 1.2 | -7 to -39.3 | OVA | C57BL/6, BALB/c and OT-II mouse models | Lymph node targeting |
| [62] | Poly (lactic-co-glycolic acid) | 100 to 200 | -22 to -25 | None | C57BL/6 melanoma mouse model | RNA and DC-based targeting |
| [63] | YSK12-MEND lipid | 180 | 5.8 | None | E.G7-OVA murine lymphoma cells | |
| [64] | Poly (lactic-co-glycolic acid) | 82.8 ± 5.7 | 27.2 ± 2.2 | Mannose and PEG | B16-F10 melanoma mouse model | Tumor-Associated Macrophage Targeting |
| [65] | Gold | 23.2 ± 2.8 | -30 ± 1.7 | Thiol-PEG-COOH, M2 peptide,thiol-siRNA | BALB/c mice with A549 human lung adenocarcinoma model | RNA + TAM targeting |
* Data not available; ** G = Generations
Nanoparticles are easy to fabricate, and have demonstrated specific targeting of DCs without eliciting undesirable immune responses [44, 48]. For passively targeting DCs present in the lymph nodes, modifying particle sizes can increase endocytotic uptake by enhancing lymph node drainage of the nanoparticle formulation [44, 60]. Sub-50 nm particles have been implemented in research in accordance with the size-dependent uptake trend observed in lymph nodes. [44, 47, 48]. However, particles with surface modifications, such as oligodeoxynucleotides like CpG, antibodies like αCD40-mAb, or model antigens like ovalbumin (OVA), have shown active and selective targeting in DCs, and thereby increased T-cell activation [44, 47, 48].
In recent work, OVA-targeted micelles have demonstrated specific caveolae-mediated internalizetion and amplification of antigen cross presentation properties. Conjugated micelles showed a 5.5-fold uptake of OVA and CpG while non-conjugated showed a 2.5-fold uptake in the DC2.4 cell line. Also, conjugated formulations exhibited increased CTL-activation via DC-targeting in C57BL/6 mice [50]. In another study involving dual targeting, DC-surface mannose receptors were used for delivery of melanoma specific antigen p-Trp2, along with the potent adjuvant, CpG. Mannose-PEG modified nanoparticles showed an increase in delivery efficacy of the antigen in B16-F10 metastatic mouse models compared to the control [51].
Nanoparticles of sub-300nm sizes have also shown applications in targeting DCs with appropriate modifications using targeting ligands [42, 43, 61]. Utilizing moieties such as CD40-mAb or OVA conjugated polymeric nanoparticles have shown increased uptake by DCs and subsequent priming of CTLs in vivo [44, 46]. However, it was recently demonstrated that surface modification is not the sole advantage that nanoparticles offer. RNA lipoplexes without surface modifications sized under 320 nm exhibited promising results in clinical trials [43].
A range of polymers and lipids have been used to formulate nanocarriers. Polymers, like PLGA modified with PEG, have shown improved biocompatibility and longer in vivo circulation times, which prevents undesirable uptake by scavenger cells, thus specifically targeting DCs [44, 51]. Lipids, too, have been determined to be efficient nanocarriers for RNA delivery to DCs, resulting in low dosage requirements and high transfection efficiencies, without the need for surface modifications [35, 63]. Fang et al. in their study coated poly (lactic-co-glycolic acid) (PLGA) nanoparticles with B16-F10 melanoma cancer cell membranes containing tumor associated antigen and homotypic binding antigen to initiate tumor-specific immune response [49]. Monophosphoryl lipid A (MPLA), an inducer of Toll-like receptor 4 (TLR-4), was incorporated onto the cancer cell membrane coated nanoparticles to increase antigen-specific immune response. Dendritic cell (DC) maturation and higher DC uptake of these particles was observed in this system. These studies potentiate the nanotechnology approach for efficient in vivo delivery of antigens and adjuvants, and successful activation of CTL responses by targeting DCs.
A novel technique for manufacturing artificial-APCs (aAPCs) for direct CTL activation was adopted by Perica et al. Iron-oxide and quantum dot nanoplatforms coated with dextran and avidin respectively were synthesized with sizes sub-100 nm. Both formulations demonstrated tumor rejection via CTL-priming. Iron-oxide dextran coated aAPCs showed dose-mediated T-cell proliferation. T-cell proliferation increased 450-fold through micro-aAPCs, while nano-aAPCs induced a 650-fold increase at equal protein concentrations. The increase in T-cell proliferation between micro and nano-aAPCs can be attributed to better lymphatic drainage in nanoparticles. Moreover, iron-oxide nano-aAPCs showed co-localization with T-cells whereas micro aAPCs did not due to low lymphatic drainage [60].
Tumor Associated Macrophage (TAM) infiltration has also been associated with tumor proliferation in several cancer types. In recent studies by Zhu S. and Conde et al, polymeric and gold nanoparticles with surface modifications were employed for selectively targeting TAMs [64, 65]. The acidic property of the tumor microenvironment was exploited by PEGylated-poly (lactic-co-glycolic acid) nanoparticles targeting mannose receptors on TAMs. 50% to 100% PEG-shedding was observed in these acid-sensitive (pH 6.8) mannose targeting nanoparticles (AS-M-NP) in vitro. Acid-sensitive PEG-shedding properties decreased undesirable uptake in vitro by 75% and showed decreased uptake by mononuclear phagocyte system (MPS) organs consisting of spleen and liver in B16-F10 melanoma mouse models.
Viral Vector-based Immunotherapy
Immunotherapy using viruses was earlier thought to be unsuccessful due to their inherent self-replicating properties, coupled with their mutating tendencies. Research on oncolytic viruses has shifted from oncolytic virotherapy to oncolytic immunotherapy. Jonathan Pol et al. published the first FDA approved study for melanoma immunotherapy involving a genetically modified herpes simplex virus, initially called Talimogene/ OncoVEXGM-CSF and later renamed Imlygic by Amgen [66, 67]. Lebel, M.E., et al., showed that subcutaneous administration of papaya mosaic virus (PapMV), an immunostimulatory agent, into B16 melanoma mouse models can curb metastasis into the lungs. Upon injection of PapMV, there was an increase in CD8+ T-cell number and a decrease in the number of myeloid suppressor cells [68]. PapMV also acts as a dendritic cell vaccine by stimulating an innate immune response through production of IFN-α.
Viral like particles (VLPs) are antigenic determinants in vaccines which lack nucleic acids. They cause the immune system to produce antibodies against viral coat proteins [69]. In a recent study by P. H. Lizotte et al., they used cowpea mosaic virus (CPMV), a plant virus that self-assembles into icosahedral shaped nanoparticles, as an immunostimulatory agent. Empty cowpea mosaic virus (eCPMV), i.e. viral particles devoid of the viral genome, was used in the study to trigger systemic immune response against multiple cancer types in mouse models. Key results of this study have been reproduced in Figure 2. In vitro studies showed that exposure to eCPMVs resulted in increased production of chemokines and cytokines. Upon inhalation, higher uptake of eCPMVs (10X) was observed in tumor-infiltrating neutrophils (TIN) than CD11b+ activated neutrophils (immunosuppressant neutrophils) in B16-F10 mice bearing tumors in lungs. There was no uptake of eCPMVs in macrophages and other cells. TINs expressed MHC II on their surface and exhibited increased amounts of CD86, which promotes antigen presentation. Intratracheal administration of eCPMV in B16F10 lung metastatic melanoma model showed a decrease in tumor growth (Figure 2D). In order to understand its immunological effects in other mice models, a 4T1 BALB/c syngeneic breast cancer model was used. In this model, primary tumors in mouse mammary pad metastasized to the lungs. The primary tumors were then excised and administered with eCPMV. The tumor occurrence in lungs of mice treated with eCPMV was delayed when compared with controls treated with PBS.
eCPMV as an immunostimulatory agent (A) External (left) and internal (right) view of eCPMV modified using Chimera (P.H.Lizotte et al.) (B) Mice with 4T1 breast tumors injected with PBS developed metastatic lung tumors and had less survival while those administered with eCPMV, intratracheally had delayed onset of tumors and also a profound extended survival. (C) Lungs of B16-F10 metastatic lung tumor mice treated with eCPMV via inhalation reduced the tumor burden over 21 days, while the lungs of those treated with PBS increased the tumor burden. (D) Mice with CT26 colon tumors responded to eCPMV by forming a necrotic area in the injected region leading to delayed tumor growth than the control group. (E) Mice implanted with ovarian carcinoma were administered with eCPMV intraperitoneally and monitored over 42 days. Those treated with eCPMV had enhanced survival with no ascites while mice injected with PBS had ascites in the abdominal area. (F) Mice survived from B16F10 melanoma tumors after treatment with eCPMV, reject when reinjected with the tumor while the naïve mice developed tumors.
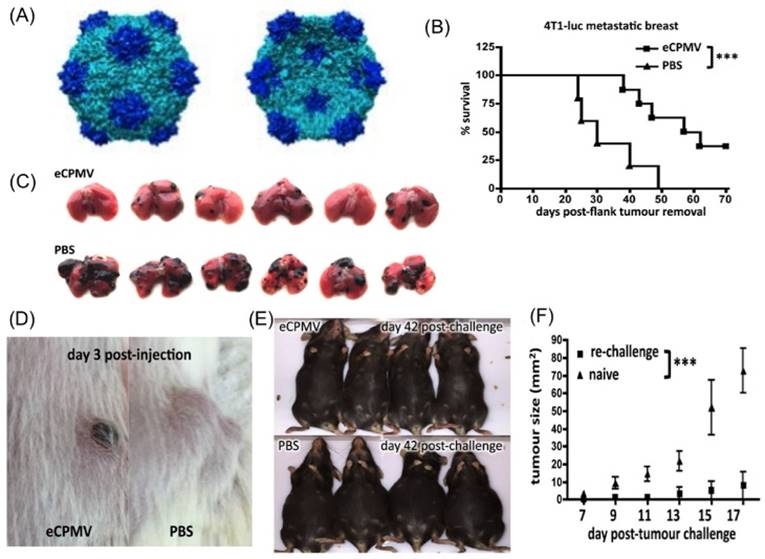
To understand the efficacy of eCPMV in different types of cancer models, an intradermal CT26 colon tumor model was injected with eCPMV, which resulted in delayed colon tumor growth. However, necrosis was also observed in the injected area as shown in Figure 2D. When eCPMVs were injected intraperitoneally into serous ovarian carcinoma mice models, significantly higher subject-survival and flank-tumor reduction was observed in the treatment group after 42-days (Figure 2E). B16 dermal melanoma mice were directly injected with eCPMV wherein 50% mice showed tumor eradication. To test the systemic immunity, the mice that survived were re-injected with B16-F10 tumor cells on their flanks 4 weeks post disappearance of primary tumors. Most of the mice showed continued resistance to B16F10 tumor cells (Figure 2F), suggesting that the systemic immunity is established. eCPMVs have been shown to bind to APCs [70], as well as to vimentin on cancer cells [71, 72] to activate neutrophils, which then recruit CTLs. This monotherapy can be incorporated as one of the treatments to treat the types of cancer mentioned above.
Immunotherapy Based on mRNA Delivery
RNA technology allows for a precise immune response by either encoding for specific antigens or by silencing mRNA, resulting in selective expression of targeted molecules [43, 63, 65]. In recent studies, competence of lipid nanocarriers via physicochemical modifications was demonstrated, and desired localization and delivery of encapsulated antigen-encoding RNA was observed [63]. pH-sensitizing modifications of nanoparticles have shown abilities such as endosomal escape in dendritic cells, increasing RNA delivery and thereby eliciting an efficacious immune response [63, 65]. Nanoparticle-mediated delivery, therefore, presents itself as a promising approach for the effective implementation of RNA-based immunotherapy.
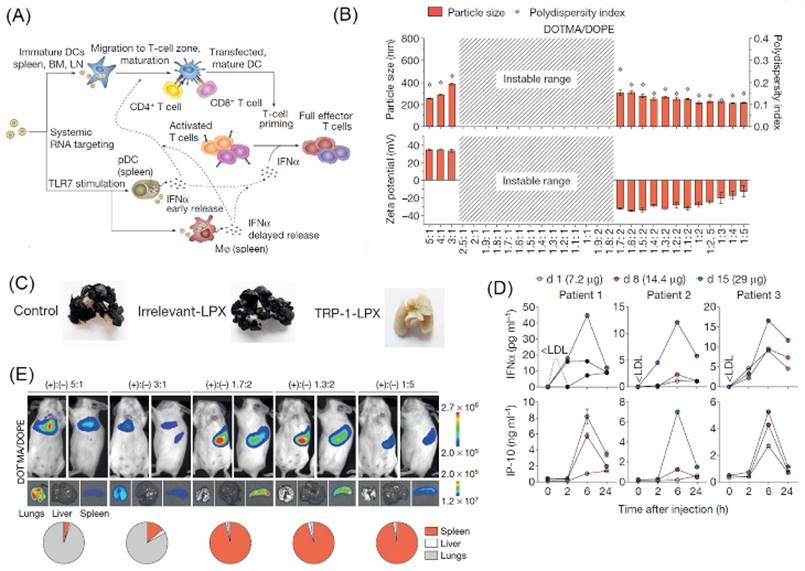
In a study by Kranz et al., the administration of a nanoparticle lipoplex formulation containing RNA encoding for tumor-specific antigens (RNA-LPX) was investigated. Analyzing the immune response elicited by targeting APCs with RNA-LPX formed the basis of their novel approach. Key results of the study have been reproduced in Figure 3. The nanoparticle comprised of cationic lipids DOTMA or DOTAP and helper lipids DOPE or cholesterol, which also contributed to its positive charge, encapsulating negatively charged RNA. A gradual decrease in the cationic content demonstrated an increase in the splenic uptake of RNA-LPX. Although RNA-LPX particles with a slight positive charge typically show instability and particle aggregation, selecting an RNA-LPX formulation with a charge ratio (negative: positive) of 1.3:2 exhibited selective targeting of the spleen. This charge ratio generated monodispersed and stable particles sized 200-320 nm, as shown in Figure 3B, which were resistant to degradation by mouse serum at body temperature. In vivo bioluminescent imaging (Figure 3E) demonstrated exclusive splenic signals for near-neutral and slightly negative RNA-LPX encoding the luciferase gene (Luc-RNA-LPX), whereas slightly positive RNA-LPX demonstrated a higher uptake in lungs rather than spleen.
Kranz et al. also demonstrated efficient cytoplasmic translation of RNA-LPX encoding influenza virus hemaglutinin (HA) in conventional DCs (cDCs) and plasmacytoid DCs (pDCs) with upregulation in activation markers. Activated cells showed a transient burst release of interferon-α (IFN-α) peaking at 6 hours after intravenous injection in mice. TLR-deficient mice exhibited significantly lower systemic IFN-α release. Antigen specific T-cell stimulation was achieved using RNA-LPX encoding for influenza virus hemagglutinin (HA-LPX). HA-LPX splenocytes were activated in vivo for functional antigen presentation. A B16-OVA lung metastasis model, which utilizes a lung cancer cell line genetically modified to express ovalbumin, was used in this study to evaluate efficacy of RNA-LPX. Tumor bearing C57BL/6 mice were inoculated with three doses of RNA-LPX encoding ovalbumin (OVA-LPX). Complete remission of metastasized lung tumors was observed in OVA-LPX immunized mice 20 days after the final dose.
Clinical studies have shown dose-dependent release of IFN-α in three patients (see Figure 3D) peaking at 6h when treated with RNA-LPX vaccines encoding four tumor antigens (NY-ESO-1, MAGE-A3, tyrosinase and TPTE). Recruitment of T cells targeting NY-ESO-1 was observed in one patient two weeks after immunization. In another patient, surgical removal of metastasized tumors followed by vaccination was shown to induce CD4+ T cells targeting NY-ESO-1 and MAGE-A3, resulting in tumor eradication after seven months. The third pre-treated patient with eight lung metastasis exhibited induction of strong NY-ESO-1 T-cell response and a weaker response against MAGE-A3. This study, therefore, exploited the physicochemical properties of non-functionalized nanoparticles to mimic a viremic pathogen intrusion and elicit T-cell immune response.
RNAi-based Immunotherapy
RNAi is the repression or downregulation of a target protein at the mRNA level. RNAi utilizes small interfering RNAs (siRNAs) or microRNAs (miRNAs), that are complementary in sequence to a region of the mRNA target. Once bound to the mRNA, the siRNAs or miRNAs interact with the cell's RNA induced silencing complex (RISC), which degrades the mRNA, preventing translation.
(A) Schematic showing IFN-α release triggered by RNA-LPX transfection via systemic delivery. (B) RNA-LPX demonstrating fair uniformity in particle sizes, and a preferred stability with negative charge ratios. (C) Complete rejection of B16-F10-Luc tumors in C57BL/6 mice by T-cell response induction against TRP-1 melanocyte-differentiation antigen on treatment with TRP-1-LPX as compared to control and irrelevant-LPX. (D) Phase I dose-escalation studies showing serum concentrations of cytokines IFN-α and IP-10 peaking at 6 hours after intravenous administration to three patients. (E) Organ localization of Luc-LPX using bioluminescence imaging in BALB/c mice after i.v. injection at different charge ratios.
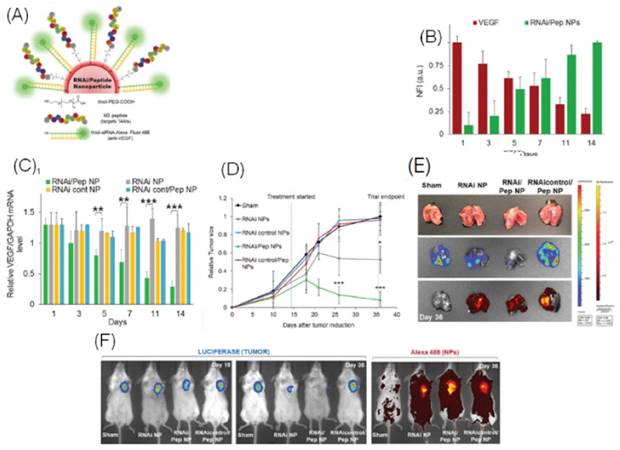
In a recent study, a nano-delivery system developed by Conde et al., transported siRNAs to both tumor-associated macrophages (TAMs) and lung cancer cells. High dual-targeting efficacy using gold nanoparticles (GNPs) was demonstrated. Key results of this study have been highlighted in Figure 4. Common non-selective strategies for targeting TAMs are inhibition of macrophage recruitment, and enhancement of macrophage tumoricidal activity. However, the M2pep-targeting approach investigated in this study selectively and preferentially bound to TAMs. As shown in Figure 4A, functionalized GNPs with vascular endothelial growth factor (VEGF) silencing RNAi with TAM-targeting peptide (M2pep) were synthesized to dually target TAMs and lung cancer cells in order to produce a synergistic effect. This RNAi-M2pep-GNP formulation exhibited a particle size and charge of 23.2 ± 2.8 nm and -30 ± 1.7 mV respectively. The siRNA:GNP and M2pep:GNP ratios of 50:1 and 30:1 respectively proved desirable for this application.
Peptide conjugated particles showed higher uptake in the perinuclear region and VEGF-silencing activity in C8 human lung adenocarcinoma cells. In BALB/c nude mice, 75% of targeted particles were found to deliver siRNA to the cytoplasm following lysosomal escape, with an 80% decrease in VEGF expression compared to controls, and a reduction in the tumor size to one-sixth of the pre-treatment size. Conde et al. also established that type I interferon or TLR immune response, a major concern in siRNA nano-delivery, did not contribute to the observed results. In addition, the formulations were found to be non-cytotoxic; however, long-term accumulation effects and excretion of particles is an area yet to be researched.
(A) Schematic diagram of gold nanoparticle conjugated with thiolated-siRNA for VEGF-knockdown and Alexa Fluor 488 for fluorescent labeling, and M2pep for selectively targeting tumor-associated macrophages. (B) Normalized Fluorescence Intensity (NFI) displays decreasing VEGF expression with increasing RNAi-M2pep-GNPs over a 14-day period. (C) RNAi-M2pep-GNP targeted formulation showing greater downregulation of VEGF as compared to RNAi-GNP and other controls over a 14-day period. (D) Graph depicts drastic reduction in lung tumor xenograft sizes in BALB/c mice treated with intratracheal administration of RNA-M2pep-GNPs as compared to controls while image (E) shows bioluminescent images for the same. (F) Bioluminescence in vivo imaging of regression of A549-luciferase-C8 human lung adenocarcinoma tumors in BALB/c nude mice and fluorescence imaging of Alexa Fluor 488 conjugated siRNA in RNAi-M2pep-GNP and controls.
Reporter Nanoparticles
Tumor heterogeneity hinders the success of existing therapies. With current procedures, we fail to understand the activity of drug delivery systems in vivo. Theranostics are a blend of therapeutics and diagnostics, primarily designed to monitor drug delivery and visualize the real-time efficacy of therapies. They are popularly used in the field of nanomedicine to track drug release patterns and nanoparticle retention in leaky vasculatures.
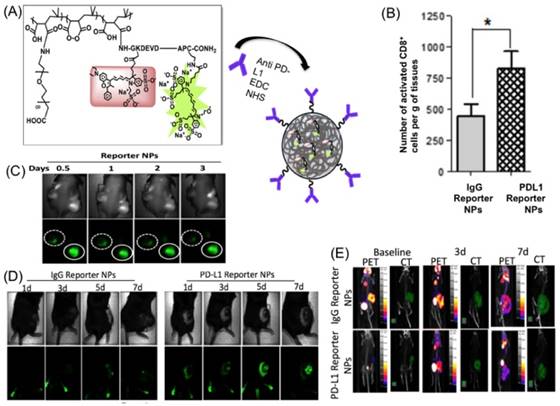
A self-reporting, or “reporter” nanoparticle, was designed by Kulkarni et al. that allows the user to see which cells of the tumor are susceptible to an immune response, and which cells are not. This novel design involves poly (isobutylene-alt-maleic anhydride) (PIMA) encapsulating an effector element, paclitaxel, and a reporter element: a Fluorescence Resonance Energy Transfer (FRET) pair, DyLight 755 (a fluorescent dye) and DyLight 766 (a quencher), conjugated to the L-amino acid sequence: GKDEVDAPC-CONH2. PEG conjugated to anti PD-L1 (programmed death ligand) antibodies were also included in the nanoparticle to allow for targeting of immune-susceptible cells. The antibodies target PD-L1 receptors on CD4+ and CD8+ T-cells. Transport of the NPs was achieved when these cells infiltrated the tumor. When Caspase-3 was activated by paclitaxel [73], it cleaved the DEVD portion of the peptide in the reporter element and blocked the quenching of the fluorescence dye, emitting a fluorescence signal.
The above mentioned reporter nanoparticles were injected into B16-F10 melanoma mouse models. IgG reporter nanoparticles were used as a control. On day 5, fluorescence signaling was pronounced in PD-L1 reporter nanoparticle treated mice compared to control mice (Figure 5C). Activated CD8+ (CTLs) were also elevated in the treatment group (Figure 5D). [18F] FDG-PET or CT imaging was performed on the mice in both the experimental and control groups. No reduction in uptake of FDG in tumor cells was observed after 7 days post-treatment in either group (Figure 5E). There was no difference between the baseline PET/CT signal and the PET/CT images taken on day 3 and day 7. FDG-PET/CT requires a wait time of at least 30 minutes [77]. Hence, a limitation of this study is the inability to monitor the real time effect of the reporter nanoparticle in vivo. Another limitation of this work is the use of fluorescent dyes, which, when translated to clinical settings, needs to be changed to radiocontrast dyes like Iodine (131I), Barium (137Ba).
Reporter nanoparticle design and in vivo testing (A) Synthesis of PD-L1 reporter nanoparticle formed by conjugating carboxy-PEG and the effector element to PIMA through self-assembly and PD-L1 antibody conjugated to PEG. (B) The graph indicates the number of activated CD8+ T-cells per gram of tissue, with a significant higher number in mice treated with PD-L1 reporter nanoparticles than the control. (C) In vivo fluorescence imaging of BALB/c nude mice with DU145 paclitaxel sensitive tumor on one flank and DU145-TR paclitaxel resistant tumor on the other flank. When injected with reporter nanoparticles, the solid circle represents the sensitive one with bright fluorescence while the resistant tumor in the dotted circle had no uptake. (D) Fluorescence images of B16-F10 melanoma mice model treated with PD-L1 reporter nanoparticles show increasing fluorescence with time while the control treated with Immunoglobulin G (IgG) has reduced fluorescence. (E) [18F] PET/CT images of B16-F10 melanoma mice treated with PD-L1 reporter nanoparticles and IgG nanoparticles taken on day 3 and day 7.
Chemoimmunotherapy
Chemoimmunotherapy is a combination of chemotherapy and immunotherapy, in which the drug-encapsulated nanoparticles have antibodies on their surface that specifically bind to the target cells. Shahbazi et al. designed undecylenic acid modified thermally hydrocarbonized porous silicon nanoparticles (UnTHCPSi NPs) containing sorafenib and the surface-conjugated with anti-CD326 antibodies (Abs), making them UnAbs [35]. The effect of UnTHCPSi NPs with and without anti-CD326 Abs, when tested on CD326 positive MCF-7 and MDA-MB-231 cells, revealed higher inhibitory activity in NPs containing anti-CD326 Abs. In cells that are CD326 negative, no difference in inhibitory activity was observed in UnTHCPSi NPs and UnAbs. This suggests that the CD326 receptor is an important target in the therapy. Antibody dependent cellular cytotoxicity (ADCC) was determined to check if Abs conjugated to the nanoparticles were functionally active. Treatment with UnAbs resulted in concentration dependent ADCC activity in MCF-7 cells, while no such difference was observed in MDA-MB-231 cells.
In another study, paclitaxel loaded 4.5G Poly (propylene-imine) dendrimers were conjugated with mAbK1 antibodies, to form mAbK1-PPI-PTX immunodendrimers. These were used to mitigate the ovarian cancer activity in OVACAR-3 and A-431 cells [52]. The mAbK1 is specific to mesothelin protein, which is aberrantly expressed in ovarian cancer cells. MTT cytotoxicity results showed that mAbK1-PPI-PTX showed twice the cytotoxicity than the free PTX and PPI-(CN)-PTX. In vivo studies in BALB/c mice injected with OVCAR-3 cells showed a significant reduction in tumor compared to the controls. Immunodendrimers with an increased half-life showed higher uptake by the tumor cells, facilitating higher drug delivery and an increase in therapeutic effects.
Alternate Targeting Strategies
Sasso et al. used PEGylated lipid nanocapsules containing lauroyl modified gemcitabine (GemC12) to reduce the number of tumor induced myeloid derived suppressor cells (MDSCs). These cells otherwise suppress the activation of cytotoxic T-cells in patients after Adoptive T-cell therapy (ACT). Higher uptake of lipid nanocapsules was observed in monocytes. Low levels of gemcitabine in the nanocapsules significantly reduced tumor immunosuppression [56]. C57BL/6 CD45.2 mice injected with EG7-OVA tumors were treated with GemC12 lipid nanocapsules on day 8, followed by ACT on day 9. Increased distribution of CD8+ and CD45.1+ cells were found in the lymph nodes when tumor cells and normal cells were cultured with OVA peptide. Gold nanoparticle (GNP) accumulation in the immune cells was used as a tool by Adam Yuh Lin et al. to stimulate an immune response. GNPs of 15 nm in size were conjugated with poly-thymidine spacer attached to triethylene glycol (TEG), which in turn is attached to CpG. TEG and Poly-T spacers enhanced the immunostimulatory response and increased the cell uptake. C57BL/6 mice implanted with B-16 OVA tumor cells, were injected with the aforementioned nanoparticle formulation (tmCpG-GNP) and % survival of these mice was significantly higher than the controls treated with PBS and free CpG [57].
Albumin nanoparticles encapsulating pyropheophorbide-a (Ppa) conjugated with TA99 mAbs were used by Chu et al. to target tumors using TINs. Mice with melanoma were injected with NPs containing TA99 specific to the gp75 antigen, which is expressed on neutrophil cell membranes. Neutrophils delivered Ppa to the tumor site and tumor growth was suppressed using photodynamic therapy [59].
Chattopadhyaya et al. developed a treatment method using cobalt oxide (CoO) nanoparticles coated with phosphonomethyliminodiacetic acid (PMIDA) and conjugated with lysate antigen. These NPs, when injected into swiss mice, showed an increased IgG response and a higher CD4+ response. These NPs activated macrophages, resulting in higher TNF-α and IFN expression. The study revealed that the immunological response was only due to the lysate antigen conjugated to the particles [58]. Zhu et al. synthesized PLGA nanoparticles, conjugated with mannose and PEG with different combinations of acid-sensitive and acid insensitive surface modifiers [64]. These nanoparticles were tested in C57BL/6 mice implanted with B16-F10 melanoma tumors. FITC conjugated PLGA was implemented for in vivo and in vitro testing. Co-localization of FITC labeled nanoparticles in macrophages was observed in the case of acid sensitive PEG conjugated PLGA, where PEG was shed in the low pH tumor environment. This system provides a path to design efficient drug delivery system to target tumor associated macrophages.
Hu et al. designed oral DNA vaccines by coating live attenuated Salmonellae with a cationic polyplex. This oral vaccine encodes for Vascular endothelial growth factor receptor 2 (VEGFR2). When a B16 melanoma tumor model was treated with this system, higher CD8+ was observed when compared with the controls [53]. These mice exhibited high concentration of cytokines such as TNF-α, IFN, and IL-12. Thus, this design showed suppression of tumor angiogenesis.
Recent research using nanoparticles for cancer immunotherapy has demonstrated the advantage of physicochemical manipulation in improving the delivery of immunostimulatory agents. In vivo studies have tested a range of particle sizes, mostly less than 300 nm, and particles with both positive and negative zeta potentials for various applications. Material composition and surface modifications have been shown to contribute significantly in selective targeting, efficient delivery and active stimulation of immune system targets [35, 41, 43-66]. Thus, these investigations, including a wide array of nanoparticles for cancer immunotherapy, substantiate the employment of nanocarriers for efficacious cancer immunotherapies.
Conclusions
Despite the incremental enhancement typically observed with chemo- and radiotherapy in cancer management, the war against cancer still continues to use some combination of these different therapeutic modalities. This explains the extremely high remission rates among advanced cancers. Over the last decade, immunotherapy has been emerging as a promising strategy to tackle heterogeneity in cancer. Cancer immunotherapy is expected to be disruptive to the field of cancer research, changing the paradigm of cancer management and moving the focus towards targeted therapy instead of maintenance therapy. The major advances in this field include the approvals of the anti-CTL4 monoclonal antibody and T-VEC, the first oncolytic viral vector-based therapy, by the FDA. Several clinical trials using immunotherapy for cancer are currently ongoing. These recent successes in cancer immunotherapy are driving great expectations, especially towards regulating the role and function of immune cells within the tumor microenvironment. Among these strategies, the use of nanoparticles for immunomodulation and viral vector-based cancer vaccines were highlighted in this review. Work highlighted in this review conclusively establishes that nanotechnology can be combined with immunotherapy to enhance the efficacy of immunogenic small molecule drugs and biologics by changing their delivery mechanism, biodistribution, and pharmacokinetics in the tumor microenvironment.
However, immunotherapy is not without its drawbacks. Like many emerging technologies, early adopters of these treatment modalities can expect a hefty price tag. Amgen has been quoted saying that its T-VEC treatment would cost on average $65,000 per treatment [67]. This would put not only a massive economic strain on the patient, but also on the healthcare system as a whole. In addition, few of the aforementioned studies demonstrated complete tumor remission, suggesting multiple rounds of treatment if and when they reach the clinic. Recently, an immunotherapy by Bristol-Myers Squibb failed phase III clinical trials. Their product, Opdivo (generically termed as 'nivolumab'), a PD-1 inhibitor, was administered as a first line of treatment to non-small cell lung cancer patients, but it failed to slow down the progression of the tumor. A nanoformulation of this treatment can result in better treatment efficacy [74, 75, 76].
Improvements in non-invasive (or minimally invasive) imaging strategies are going to be vital to guide the delivery of immunotherapies and monitor the associated treatment responses. Molecular imaging modalities, in combination with immunostimulation (in a single nanometer-sized platform), would enable researchers to obtain comprehensive, real-time, information regarding the biological signaling and crosstalk within the various components that make-up the complex tumor microenvironment. The development of non-toxic, biocompatible, biodegradable nanotechnology-based delivery systems that can combine imaging probes and specifically target immune cells will usher in a new era of next generation nano-immunotheranostics. These nano-immunotheranostics will help personalized treatment planning, which will result in better treatment outcomes with little or no side effects. Such effective treatment outcomes with reduced morbidities will aid the quick translation of engineered nano-immunotheranostics into the clinic.
Abbreviations
FDA : Food and Drug Administration; CTLA-4: Cytotoxic T-Lymphocyte Associated Protein 4; DDS: Drug Delivery System; PEG: poly-ethylene-glycol; APCs: Antigen-Presenting Cells; DCs: Dendritic Cells; mAbs : monoclonal antibodies; CTL: Cytotoxic T Lymphocyte; PIMA: poly isobutylene-alt-maleic anhydride; FRET: Fluorescence Resonance Energy Transfer; eCPMV: empty Cowpea Mosaic Virus; OVA: Ovalbumin; PLGA: poly (lactic-co-glycolic acid); MPLA: Monophosphoryl lipid A; TAM: Tumor Associated Macrophage; MPS: Mononuclear Phagocyte System; PapMV: Papaya Mosaic Virus; TIN: Tumor-Infiltrating Neutrophils; MHC II: Major Histocompatibility Complex II; mRNA: messenger Ribonucleic acid; DOTMA: 1,2-di-O-octadecenyl-3-trimethylammonium propane; DOTAP: 1,2-dioleoyl-3-trimethylammonium-propane; LPX: Lipoplex; HA: hemaglutinin; IFN-α: Interferon-α; siRNA: small interfering RNA; miRNA: micro RNA; RISC: RNA induced silencing complex; GNP: Gold nanoparticles; VEGF: Vascular Endothelial Growth Factor; RNAi: RNA interference; ADCC: Antibody Dependent Cellular Cytotoxicity; PPI: Poly (propylene-imine); PTX: Paclitaxel; MDSC: Myeloid derived suppressor cells; ACT: Adoptive cell therapy; TEG: Triethylene glycol; PMIDA: phosphonomethyliminodiacetic acid.
Acknowledgements
The authors acknowledge funding from the Office of Cancer Nanotechnology Research at the National Cancer Institute, a part of the National Institutes of Health (Grant #R00CA153948).
Author Contributions
P.V., A.G., B.P., R.J. and P.R. designed and wrote this article.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Goldman A, Kulkarni A, Kohandel M, Pandey P, Rao P, Natarajan SK. et al. Rationally designed 2-in-1 nanoparticles can overcome adaptive resistance in cancer. ACS nano. 2016;10:5823-34
2. Shao K, Singha S, Clemente-Casares X, Tsai S, Yang Y, Santamaria P. Nanoparticle-based immunotherapy for cancer. ACS nano. 2014;9:16-30
3. Restifo NP, Smyth MJ, Snyder A. Acquired resistance to immunotherapy and future challenges. Nature Reviews Cancer. 2016;16:121-6
4. Mendes F, Domingues C, Rodrigues-Santos P, Abrantes AM, Gonçalves AC, Estrela J. et al. The role of immune system exhaustion on cancer cell escape and anti-tumor immune induction after irradiation. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer. 2016;1865:168-75
5. Schreiber R.D, Old L.J, Smyth M.J. Cancer immunoediting: Integrating immunity's roles in cancer suppression and promotion. Science. 2011;331:1565-1570
6. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. cell. 2011;144:646-74
7. Tran E, Robbins PF, Lu Y-C, Prickett TD, Gartner JJ, Jia L. et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. New England Journal of Medicine. 2016;375:2255-62
8. Morvan MG, Lanier LL. NK cells and cancer: you can teach innate cells new tricks. Nature Reviews Cancer. 2016;16:7-19
9. Pitt JM, André F, Amigorena S, Soria J-C, Eggermont A, Kroemer G. et al. Dendritic cell-derived exosomes for cancer therapy. Journal of Clinical Investigation. 2016;126:1224
10. Li Y, Yuan J, Yang Q, Cao W, Zhou X, Xie Y. et al. Immunoliposome co-delivery of bufalin and anti-CD40 antibody adjuvant induces synergetic therapeutic efficacy against melanoma. International journal of nanomedicine. 2014;9:5683
11. Tao Y, Ju E, Liu Z, Dong K, Ren J, Qu X. Engineered, self-assembled near-infrared photothermal agents for combined tumor immunotherapy and chemo-photothermal therapy. Biomaterials. 2014;35:6646-56
12. Sarkar S, Pradhan A, Das SK, Emdad L, Sarkar D, Pellecchia M. et al. Novel therapy of prostate cancer employing a combination of viral-based immunotherapy and a small molecule BH3 mimetic. Oncoimmunology. 2016;5:e1078059
13. Neves H, Kwok HF. Recent advances in the field of anti-cancer immunotherapy. BBA clinical. 2015;3:280-8
14. Romano E, Kusio-Kobialka M, Foukas PG, Baumgaertner P, Meyer C, Ballabeni P. et al. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proceedings of the National Academy of Sciences. 2015;112:6140-5
15. Cheever MA, Higano CS. PROVENGE (Sipuleucel-T) in prostate cancer: the first FDA-approved therapeutic cancer vaccine. Clinical Cancer Research. 2011;17:3520-6
16. Di Lorenzo G, Ferro M, Buonerba C. Sipuleucel-T (Provenge®) for castration-resistant prostate cancer. BJU international. 2012:110
17. Sartor O. SIPULEUCEL-T (PROVENGE®) FOR CASTRATION-RESISTANT PROSTATE CANCER. BJU international. 2012:110
18. Fellner C. Ipilimumab (Yervoy) prolongs survival in advanced melanoma. Drug Forecast. 2012;37:503-30
19. FDA: Maryland USA. Nivolumab (Opdivo) for Hodgkin Lymphoma. http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm501412.htm
20. FDA: Maryland USA. FDA approves Keytruda for advanced non-small cell lung cancer Revised 15 September 2016. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm465444.htm
21. FDA: Maryland, USA. FDA approves Blincyto to treat a rare form of acute lymphoblastic leukemia: First anti-CD19 drug to receive agency approval. Revised 10 December 2014. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm425549.htm
22. FDA: Maryland USA. FDA Approval for Ipilimumab; Revised 15 September 2016. https://www.cancer.gov/about-cancer/treatment/drugs/fda-ipilimumab
23. FDA: Maryland, USA. FDA News Release: FDA Approves a Cellular Immunotherapy for Men with Advanced Prostate Cancer; Revised 6 May 2013. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm210174.htm
24. FDA: Maryland USA. Ovarian Cancer and Modern Immunotherapy: Regulatory Strategies for Drug Development; Revised 15 September 2016. http://www.fda.gov/downloads/drugs/newsevents/ucm464305.pdf
25. Clinicaltrials.gov. Study of Copanlisib in Combination With Standard Immunochemotherapy in Relapsed Indolent Non-Hodgkin's Lymphoma (iNHL) (CHRONOS-4); Revised 29 November 2016. https://clinicaltrials.gov/ct2/show/NCT02626455?term=Copanlisib&rank=3
26. Clinicaltrials.gov. Treatment of PCNSL With R-IDARAM and Intrathecal Immunochemotherapy; Revised 15 September 2016. https://clinicaltrials.gov/ct2/show/NCT02657785
27. Clinicaltrials.gov. Avelumab in Non-Small Cell Lung Cancer (JAVELIN Lung 200); Revised 17 October 2016. https://clinicaltrials.gov/ct2/show/NCT02395172
28. Clinicaltrials.gov. Bruton's Tyrosine Kinase (BTK) Inhibition in B-cell Lymphomas (BIBLOS); Revised 15 September 2016. https://clinicaltrials.gov/ct2/show/NCT02055924
29. Goldberg MS. Immunoengineering: how nanotechnology can enhance cancer immunotherapy. Cell. 2015;161:201-4
30. Kodali V, Littke MH, Tilton SC, Teeguarden JG, Shi L, Frevert CW. et al. Dysregulation of macrophage activation profiles by engineered nanoparticles. ACS nano. 2013;7:6997-7010
31. Chen C, Wang H. Biomedical applications and toxicology of carbon nanomaterials. John Wiley & Sons. 2016
32. Mashinchian O, Johari-Ahar M, Ghaemi B, Rashidi M, Barar J, Omidi Y. Impacts of quantum dots in molecular detection and bioimaging of cancer. Bioimpacts. 2014;4:149
33. Shargh VH, Hondermarck H, Liang M. Antibody-targeted biodegradable nanoparticles for cancer therapy. Nanomedicine. 2016;11:63-79
34. Almeida JPM, Figueroa ER, Drezek RA. Gold nanoparticle mediated cancer immunotherapy. Nanomedicine: Nanotechnology, Biology and Medicine. 2014;10:503-14
35. Shahbazi M-A, Shrestha N, Mäkilä E, Araújo F, Correia A, Ramos T. et al. A prospective cancer chemo-immunotherapy approach mediated by synergistic CD326 targeted porous silicon nanovectors. Nano Research. 2015;8:1505-21
36. Saleh T, Shojaosadati SA. Multifunctional nanoparticles for cancer immunotherapy. Human vaccines & immunotherapeutics. 2016;12:1863-75
37. Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M. et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nature nanotechnology. 2011;6:815-23
38. Agarwal R, Singh V, Jurney P, Shi L, Sreenivasan S, Roy K. Mammalian cells preferentially internalize hydrogel nanodiscs over nanorods and use shape-specific uptake mechanisms. Proceedings of the National Academy of Sciences. 2013;110:17247-52
39. Sun B, Ji Z, Liao Y-P, Wang M, Wang X, Dong J. et al. Engineering an effective immune adjuvant by designed control of shape and crystallinity of aluminum oxyhydroxide nanoparticles. ACS nano. 2013;7:10834-49
40. Carter T, Mulholland P, Chester K. Antibody-targeted nanoparticles for cancer treatment. Immunotherapy. 2016;8:941-58
41. Kulkarni A, Rao P, Natarajan S, Goldman A, Sabbisetti VS, Khater Y. et al. Reporter nanoparticle that monitors its anticancer efficacy in real time. Proceedings of the National Academy of Sciences. 2016;113:E2104-E13
42. Conniot J, Silva JM, Fernandes JG, Silva LC, Gaspar R, Brocchini S. et al. Cancer immunotherapy: nanodelivery approaches for immune cell targeting and tracking. Cancer Nanotheranostics: What Have We Learned So Far?. 2016:68
43. Kranz LM, Diken M, Haas H, Kreiter S, Loquai C, Reuter KC. et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature. 2016
44. Rosalia RA, Cruz LJ, van Duikeren S, Tromp AT, Silva AL, Jiskoot W. et al. CD40-targeted dendritic cell delivery of PLGA-nanoparticle vaccines induce potent anti-tumor responses. Biomaterials. 2015;40:88-97
45. Qian Y, Jin H, Qiao S, Dai Y, Huang C, Lu L. et al. Targeting dendritic cells in lymph node with an antigen peptide-based nanovaccine for cancer immunotherapy. Biomaterials. 2016;98:171-83
46. Xiang J, Xu L, Gong H, Zhu W, Wang C, Xu J. et al. Antigen-loaded upconversion nanoparticles for dendritic cell stimulation, tracking, and vaccination in dendritic cell-based immunotherapy. Acs Nano. 2015;9:6401-11
47. Jeanbart L, Ballester M, De Titta A, Corthésy P, Romero P, Hubbell JA. et al. Enhancing efficacy of anticancer vaccines by targeted delivery to tumor-draining lymph nodes. Cancer immunology research. 2014;2:436-47
48. Thomas SN, Vokali E, Lund AW, Hubbell JA, Swartz MA. Targeting the tumor-draining lymph node with adjuvanted nanoparticles reshapes the anti-tumor immune response. Biomaterials. 2014;35:814-24
49. Fang RH, Hu C-MJ, Luk BT, Gao W, Copp JA, Tai Y. et al. Cancer cell membrane-coated nanoparticles for anticancer vaccination and drug delivery. Nano letters. 2014;14:2181-8
50. Wilson JT, Keller S, Manganiello MJ, Cheng C, Lee C-C, Opara C. et al. pH-Responsive nanoparticle vaccines for dual-delivery of antigens and immunostimulatory oligonucleotides. ACS nano. 2013;7:3912-25
51. Xu Z, Ramishetti S, Tseng Y-C, Guo S, Wang Y, Huang L. Multifunctional nanoparticles co-delivering Trp2 peptide and CpG adjuvant induce potent cytotoxic T-lymphocyte response against melanoma and its lung metastasis. Journal of Controlled Release. 2013;172:259-65
52. Jain NK, Tare MS, Mishra V, Tripathi PK. The development, characterization and in vivo anti-ovarian cancer activity of poly (propylene imine)(PPI)-antibody conjugates containing encapsulated paclitaxel. Nanomedicine: Nanotechnology, Biology and Medicine. 2015;11:207-18
53. Hu Q, Wu M, Fang C, Cheng C, Zhao M, Fang W. et al. Engineering nanoparticle-coated bacteria as oral DNA vaccines for cancer immunotherapy. Nano letters. 2015;15:2732-9
54. Lizotte P, Wen A, Sheen M, Fields J, Rojanasopondist P, Steinmetz N. et al. In situ vaccination with cowpea mosaic virus nanoparticles suppresses metastatic cancer. Nature nanotechnology. 2016;11:295-303
55. Ma X, Hui H, Jin Y, Dong D, Liang X, Yang X. et al. Enhanced immunotherapy of SM5-1 in hepatocellular carcinoma by conjugating with gold nanoparticles and its in vivo bioluminescence tomographic evaluation. Biomaterials. 2016;87:46-56
56. Sasso MS, Lollo G, Pitorre M, Solito S, Pinton L, Valpione S. et al. Low dose gemcitabine-loaded lipid nanocapsules target monocytic myeloid-derived suppressor cells and potentiate cancer immunotherapy. Biomaterials. 2016;96:47-62
57. Lin AY, Almeida JPM, Bear A, Liu N, Luo L, Foster AE. et al. Gold nanoparticle delivery of modified CpG stimulates macrophages and inhibits tumor growth for enhanced immunotherapy. PLoS One. 2013;8:e63550
58. Chattopadhyay S, Dash SK, Mandal D, Das B, Tripathy S, Dey A. et al. Metal based nanoparticles as cancer antigen delivery vehicles for macrophage based antitumor vaccine. Vaccine. 2016;34:957-67
59. Chu D, Zhao Q, Yu J, Zhang F, Zhang H, Wang Z. Nanoparticle targeting of neutrophils for improved cancer immunotherapy. Advanced healthcare materials. 2016;5:1088-93
60. Perica K, Medero ADL, Durai M, Chiu YL, Bieler JG, Sibener L. et al. Nanoscale artificial antigen presenting cells for T cell immunotherapy. Nanomedicine: Nanotechnology, Biology and Medicine. 2013;10:119-29
61. Mueller SN, Tian S, DeSimone JM. Rapid and persistent delivery of antigen by lymph node targeting PRINT nanoparticle vaccine carrier to promote humoral immunity. Molecular pharmaceutics. 2015;12:1356-65
62. Heo MB, Cho MY, Lim YT. Polymer nanoparticles for enhanced immune response: combined delivery of tumor antigen and small interference RNA for immunosuppressive gene to dendritic cells. Acta biomaterialia. 2014;10:2169-76
63. Warashina S, Nakamura T, Sato Y, Fujiwara Y, Hyodo M, Hatakeyama H. et al. A lipid nanoparticle for the efficient delivery of siRNA to dendritic cells. Journal of Controlled Release. 2016;225:183-91
64. Zhu S, Niu M, O'Mary H, Cui Z. Targeting of tumor-associated macrophages made possible by PEG-sheddable, mannose-modified nanoparticles. Molecular pharmaceutics. 2013;10:3525-30
65. Conde J, Bao C, Tan Y, Cui D, Edelman ER, Azevedo HS. et al. Dual Targeted Immunotherapy via In Vivo Delivery of Biohybrid RNAi-Peptide Nanoparticles to Tumor-Associated Macrophages and Cancer Cells. Advanced functional materials. 2015;25:4183-94
66. Pol J, Kroemer G, Galluzzi L. First oncolytic virus approved for melanoma immunotherapy. Taylor & Francis. 2016
67. Dangi S. Cost Questions Will Follow the Approval of Amgen's Oncolytic Viral Treatment, Imlygic - American Journal of Managed Care. Revised 28 October 2015. www.ajmc.com/newsroom/cost-questions-will-follow-the-approval-of-amgens-oncolytic-viral-treatment-imlygic
68. Lebel M-È, Chartrand K, Tarrab E, Savard P, Leclerc D, Lamarre A. Potentiating cancer immunotherapy using papaya mosaic virus-derived nanoparticles. Nano letters. 2016;16:1826-32
69. Huber B, Schellenbacher C, Jindra C, Fink D, Shafti-Keramat S, Kirnbauer R. A chimeric 18L1-45RG1 virus-like particle vaccine cross-protects against oncogenic alpha-7 human papillomavirus types. PloS one. 2015;10:e0120152
70. Gonzalez MJ, Plummer EM, Rae CS, Manchester M. Interaction of Cowpea mosaic virus (CPMV) nanoparticles with antigen presenting cells in vitro and in vivo. PloS one. 2009;4:e7981
71. Steinmetz NF, Cho C-F, Ablack A, Lewis JD, Manchester M. Cowpea mosaic virus nanoparticles target surface vimentin on cancer cells. Nanomedicine. 2011;6:351-64
72. Satelli A, Li S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cellular and molecular life sciences. 2011;68:3033-46
73. Oyaizu H, Adachi Y, Taketani S, Tokunaga R, Fukuhara S, Ikehara S. A crucial role of caspase 3 and caspase 8 in paclitaxel-induced apoptosis. Molecular Cell Biology Research Communications. 1999;2:36-41
74. Hellmann M, Gettinger S, Goldman J, Brahmer J, Borghaei H, Chow L. et al. CheckMate 012: safety and efficacy of first-line (1L) nivolumab (nivo; N) and ipilimumab (ipi; I) in advanced (adv) NSCLC. J Clin Oncol. 2016;34:abstr3001
75. Horn L, Brahmer J, Reck M, Borghaei H, Spigel D, Steins M. et al. Phase 3, randomized trial (CheckMate 057) of nivolumab (NIVO) vs docetaxel (DOC) in advanced non-squamous (non-SQ) non-small cell lung cancer (NSCLC): Subgroup analyses and patient reported outcomes (PROs). EUROPEAN JOURNAL OF CANCER. 2015:S599
76. Ferris R, Blumenschein G, Fayette J, Guigay J, Colevas A, Licitra L. et al. Further evaluations of nivolumab (nivo) versus investigator's choice (IC) chemotherapy for recurrent or metastatic (R/M) squamous cell carcinoma of the head and neck (SCCHN): CheckMate 141. J Clin Oncol. 2016:34
77. Mayo Clinic. Tests and Procedures: What You Can Expect; Revised 25 March 2015. http://www.mayoclinic.org/tests-procedures/ct-scan/basics/what-you-can-expect/prc-20014610
Author Biography
Praveena Velpurisiva received her bachelor degree in Industrial Biotechnology from SASTRA University of India in 2012. She did her Masters in Biological Sciences with a major in Biotechnology at University of Massachusetts at Lowell in 2016. She is currently pursuing her PhD under the guidance of Dr.Prakash Rai in the Department of Biomedical Engineering and Biotechnology at University of Massachusetts , Lowell.
 Dr. Prakash Rai is an assistant professor at the University of Massachusetts, Lowell in the department of chemical engineering. He received his BS in chemical engineering from the University of Mumbai, India in 2003. He obtained his MS and PhD degrees in chemical and biological engineering from Rensselaer Polytechnic Institute, Troy, NY in May and December of 2007 respectively. He is the author of more than 20 journal papers and one book chapter. His current research interests include nanotechnology, photodynamic therapy, fluorescence-based contrast agents, optical imaging, image-guided drug delivery and theranostics. He is a member of the American Institute of Chemical Engineers, American Physical Society, American Chemical Society, and the American Association of Cancer Research.
Dr. Prakash Rai is an assistant professor at the University of Massachusetts, Lowell in the department of chemical engineering. He received his BS in chemical engineering from the University of Mumbai, India in 2003. He obtained his MS and PhD degrees in chemical and biological engineering from Rensselaer Polytechnic Institute, Troy, NY in May and December of 2007 respectively. He is the author of more than 20 journal papers and one book chapter. His current research interests include nanotechnology, photodynamic therapy, fluorescence-based contrast agents, optical imaging, image-guided drug delivery and theranostics. He is a member of the American Institute of Chemical Engineers, American Physical Society, American Chemical Society, and the American Association of Cancer Research.
![]() Corresponding author: Prakash_Raiedu; Tel: +1-978-934-4971
Corresponding author: Prakash_Raiedu; Tel: +1-978-934-4971