J Biomed 2017; 2:94-100. doi:10.7150/jbm.19834 This volume Cite
Review
Melanoma from Molecular Pathways to Clinical Treatment: An Up to Date Review
Christoforos Kosmidis1, Sofia Baka2, Konstantinos Sapalidis1, Nick Mixalopoulos1, Stefanos Atmatzidis1, Harilaos Koulouris1, George Anthimidis3, Nick Varsamis3, Paul Zarogoulidis4, Yan-Gao Man5, Eleni Georgakoudi1, Stelios Mantalovas1, George Koimtzis1, Alexandros Tsakalidis1, Isaac Kesisoglou1 ![]()
1. 3rd Surgical Department, "AHEPA" University General Hospital, Aristotle University of Thessaloniki;
2. Oncology Department, Interbalkan European Medical Center;
3. Surgery Department, Interbalkan European Medical Center;
4. Pulmonary Department-Oncology Unit, “G. Papanikolaou” General Hospital, Aristotle University of Thessaloniki, Thessaloniki, Greece;
5. Laboratory of Proteomics and Protein Sciences, Veterans Affair Health System, Baltimore, MD, USA.
Received 2017-2-27; Accepted 2017-5-3; Published 2017-5-25
Abstract

Skin cancer is divided to melanoma and non-melanoma. Melanoma occurs due to deregulation of melanocytes. Melanoma is frequent in Caucasian population. During the last decade there is an increase in melanoma and in many cases there is a diagnostic challenge. Moreover; choosing the right treatment is a challenge for many patients. Malignant melanoma is known to be a lethal disease due to its aggressive capacity for metastasis and resistance to therapy. In the last decade, considerable efforts have gone into development of an effective immunotherapy for treatment of melanoma. The combination of macrophage activation and other treatments such as immunotherapy, surgery, chemotherapy, and radiotherapy, may provide an effective and comprehensive anti-melanoma strategy.
Keywords: Melanoma, immunotherapy, molecular pathways.
Introduction
Melanoma represents less than 5% of all skin cancers, however; it is responsible for the majority of skin cancer-related deaths [1]. It has been observed that Malignant melanoma tends to metastasize due to its aggressive capacity and also due to resistance to therapy [2]. Melanoma is a tumor that is recognized by the immunological system and mechanisms leading to an anti-tumor immune response of potential value in the adjuvant setting. Due to this fact treatment of melanoma is mostly based in the development of immunotherapy for treatment of metastatic melanoma [3]. In specific this observation has motivated investigation of interactions between melanoma and immune cells the consequently translation of this effect into several clinical strategies. The main mechanism includes phagocytosis of macrophages which play a critical role in nonspecific defense (innate immunity) and, moreover; they have a role as antigen presenters which also help initiate specific defense mechanisms (adaptive immunity) by recruiting other immune cells such as lymphocytes. It is known that macrophages are an important component of the innate immunity against tumors and they are attracted by locally secreted chemokines [4]. The mechanism of action is based on the activation of macrophages to defend against tumors by direct tumor cytotoxicity and consequently they secrete cytokines which recruit secondary immune cells, presenting antigen to T cells [5-7]. Do date surgery is considered the most definitive treatment for early-stage melanoma, but it is rarely curative for the advanced stages of melanomas [8]. Currently immunotherapy is the best treatment, however; before this treatment modality, chemotherapy used to be the treatment of choice for metastatic melanoma and it was based on 2 FDA-approved drugs: fotemustine, dacarbazine [9]. In recent years, advances in the use of immunotherapy and targeted therapy have increased the survival benefit of patients. It has been observed that immunotherapy has the ability to enhance the duration of objective responses and provide extended prolongation of patients' survival [10].
Current immunotherapy is based on immune checkpoint inhibitors targeting CTLA4 and, in the last 5 years in PD1/PDL1 interaction [11]. Furthermore; novel targeted therapies with MAPK pathway kinase inhibitors (KIs) have been developed thanks to the discovery that BRAF and NRAS mutations which are among the major oncogenic drivers of melanoma proliferation and survival [12]. In the last decade cell invasion is considered a therapeutic intervention and stimulated an era of intense research. As a result several regulators of melanoma invasiveness have been discovered. In the last decade the identification of genetic drivers of melanoma cell which regulate proliferation and survival -such as BRAF and other activators of the MAP-kinase pathway- along with the recent development of immunotherapy has taken away in many cases the invasive treatment of melanoma as an option. However, unfortunately the mechanisms of tumor cells have the ability to resist to immunotherapy and patients who relapse with acquired resistance to BRAF and MEK inhibitors often present with melanomas that display a much more aggressive and invasive phenotype [13, 14]. Moreover; it has been observed that not every patient responds to immunotherapy and different phenotypes have been observed to be linked to innate resistance containing signatures linked to an invasive phenotype [15]. Therefore, since there cases of resistant and aggressive melanoma phenotypes or resistance to therapy occurs during treatment targeted or invasive treatment should be considered. It has been observed that acquired resistance to BRAF inhibitors is usually characterized by reactivation of the MAPK pathway [16]. There are post-transcriptional adaptive changes which induce the development of drug resistance, these are mostly mechanisms involving non-coding RNAs [17]. In the current mini review we will include the most important molecular pathways and up-to-date treatment modalities in short.
Molecular Pathways
It has been observed that drugs targeting the MAPK pathway and immune checkpoint inhibitors are the best treatments which have demonstrated effectiveness in melanoma. However, until now these treatment modalities have not been compared directly to each other. To date available data suggest that the combination of BRAF and MEK inhibitors are the most effective treatment strategy [18]. Currently melanoma is classified according to oncogenic chromosomal aberrations, correlated with the site of the tumor and pattern of proliferation, into 4 distinct groups: non-CSD, acral (located on palms, soles and nail beds), mucosal (on mucous membrane) melanoma and Chronic Sun-Damage (CSD). It has been observed that Non-CSD melanoma has a 75% frequency of v-raf murine sarcoma viral oncogene homologue B1 (BRAF) mutations. Other gene abnormalities were, on the other hand, encountered in the other groups, such as c-Kit [19], cyclin-dependent kinase 4 (CDK4) and cyclin D1 (CCDN1) [20, 21]. There is a previous classification system, the Clark's classification namely, which has proposed the stepwise theory of progression of melanoma [9] which has been adapted by many other systems [22-24]. Another theory of melanoma development is the Bishop theory [25]. In this theory it is explained that cutaneous melanoma occurs mainly due to DNA damage involving the region of cyclin dependent kinase inhibitor 2A gene (CDKN2A), as a result of exposure to excessive amounts of ultraviolet (UV) radiation from sunburn in early stages of life [25]. However, to date the genetic mechanism and the precise effect of the UV radiation in the development of cutaneous melanoma has not been fully understood. UV radiation stimulates DNA mutations and causes local damage by several mechanisms and induces apoptosis through a production of growth factors from skin cells [26]. Moreover; ultra-violate radiation is known to reduce the immunity and stimulates formation of reactive oxygen species in melanocytes locally [27]. It is known that one of the pathways involved in cutaneous cancer is mediated through CDKN2A which is a gene associated with melanoma dysregulation through its pro-oncogenic products p16INK4a (Cyclin-Dependent Kinase Inhibitor P16) and p14ARF (P14 alternate open reading frame) [28]. Furthermore; p16INK4a inhibits CDK4 and CDK6, and induces phosphorylation of the protein retinoblastoma (Rb) and liberating the transcription factor E2F which consequently result in cell cycle progression [29]. On the other hand; neuroblastoma RAS Viral Oncogene Homolog (NRAS) mutation is known to activate BRAF which results in an anarchic melanocyte growth [30, 31]. Several molecular pathways are known to be dysregulated in melanoma. It has been observed that in the MAPK pathway, the most commonly mutated proteins include RAS and RAF. It has been observed that this pathway when is activated by upstream signaling cascades [such as growth factor receptors and G-protein coupled receptors (GPCRs)], leads to cell proliferation, differentiation and migration. It has been observed in this pathway that a single-base missense transversion causes the replacement of valine with glutamic acid at amino acid residue 600 in BRAF that is detected in about 85% of nevi and melanoma [32, 33]. BRAF is a serine/threonine kinase that is part of the MAPK signaling cascade found downstream of RAS; it is known activate MEK by phosphorylation, which in turn activates ERK also by phosphorylation.
It has been observed that mutated BRAF not only upregulates its own kinase activity, but also that of MEK and ERK, which in turn promotes cell proliferation [34]. It has been observed that in addition to mutated BRAF, mutated NRAS accounts for about 20% of melanoma cases [35]. In a recent study it was presented in uveal melanoma a mutation in the GTP binding region of the G_ subunit (Q209L) blocks the cleavage of GTP to GDP and deregulates both MAPK and PI3K/AKT pathways [35].
Moreover; it has been previously presented that transgenic mice harboring this mutated G_ subunit have increased skin lesions [36]. It has been observed that ectopic expression of metabotropic glutamate receptor 1 (GRM1) in melanocytes is sufficient to induce spontaneous melanoma development in vivo and transformation in vitro [37, 38]. Moreover; the ectopic expression of GRM1 mediated melanoma agenesis is independent of the genotype of either BRAF or NRAS [39]. Until now the mechanism by which the aberrant expression of glutamate receptor 1 in melanocytes leads to oncogenesis remains unknown. Glutamate receptor can be subdivided into three groups, group I, II, and III based on sequence homologies. It is known that through the binding of the natural ligand, glutamate, to the receptor leads to phospholipase C (PLC) activation via G-protein subunits G_q/G_11 [40]. In turn it induces the cleavage of phosphatidylinositol-4, 5-diphosphate (PIP2) to the production of two second messengers, inositol 1,4,5-triphosphate (IP3) and diacyl glycerol (DAG) [41]. Inositol 1,4,5-triphosphate then diffuses into the cytosol while DAG is connected to the membrane. Furthermore; inositol 1,4,5-triphosphate when connected to cytosol results in the interaction with the endoplasmic reticulum leading to an increase in calcium concentration within the cytosol. Moreover; it has been observed that elevated calcium levels and interaction of the membrane bound DAG with protein kinase C (PKC), activates PKC's kinase activity, which is known to regulate the cellular activity of MAPK [41]. The Groups II and III of glutamate receptor membranes are coupled to the intracellular G-protein subunit, these mediate the inhibition of adenylyl cyclase [40, 41]. Consequently the activation of group II or III glutamate receptor membranes reduce cAMP formation [40, 41]. Calcium and DAG activate protein kinase C, which, in turn, activates the MAPK and PI3K/AKT pathways which allow the upregulation of cell proliferation and anti-apoptosis signals [42]. Moreover; it has been observed that activated PI3K/AKT pathway functions in tumor cell by regulating; survival, angiogenesis and epithelial mesenchymal transition (EMT). It is known that the MAPK pathway plays important roles in melanoma pathogenesis [40]. In a previous study melan-A cells, showed increased cellular proliferation upon introduction of exogenous mutated BRAF (V600E), however no tumor formed [43]. Furthermore; it was observed that introduction of exogenous glutamate receptor membranes 1 into melan-A cells resulted in cell transformation in vitro and robust tumor formation in vivo [44]. In another study in a recently published study in an in vivo mouse model with conditioned mutated BRAF (V600E), glutamate receptor membrane 1 expression was detected after mutated BRAF expression was activated, however, no tumor was detected in these mice up to 17 months of age. These results suggested that the expression of mutated BRAF and GRM1 is critical in the determination of future tumorigenesis. There are also other drivers which regulate melanoma tumorigenesis. Moreover; the PI3K/AKT pathway also can contribute to melanoma tumorigenesis through mutations or loss in PTEN and dysregulation in expression of AKT, which positively regulates the G1/S phase progression in cell cycle, suppresses apoptosis and promotes cellular survival.
Previously in a mutated BRAF mouse model, ablation of PTEN led to melanoma development [45], suggesting the PI3K/AKT signaling cascade is one of the critical players in melanoma turmorigenesis. Moreover; mutations in cyclin-dependent kinase inhibitor 2A (CDKN2A) have been observed to be common among human cancers including melanoma. It is known to encode two proteins; a) p16INK4a and b) p14ARF. P16 binds to cyclin-dependent kinases 4 and 6 (CDK4 and CDK6) and inhibits phosphorylation of Rb, which remains associated with the transcription factor, E2F. Alternate reading frame protein (ARF) complexes with mouse double minute 2 homolog 2 (MDM2), an E3 ubiquitin ligase that regulates the stability of p53 and therefore suppressing tumor growth, it is also involved in the immune response, by modulating the tumor environment [46]. Therefore, mutation(s) in alternate reading frame protein dysregulate p53 function and play a role in tumor immune evasion. It has been observed that c 4 (CDK4) gene amplification, is an independent oncogene, and is more common in acral and mucosal melanoma than the CSID and NCSID cutaneous melanomas [20, 47]. It has been observed that mutations or deletions of PTEN are concurrent with mutations in BRAF but not in N-RAS [20]. It has been previously suggested that N-RAS activates both the PI3K and MAPK pathway, while BRAF only activates the latter. This theory indicates that in melanoma pathogenesis and somatic mutations activate one pathway but require another event to activate other pathways [20].
VEGF; Vascular Endothelial Growth Factor, FGF; Fibrotic Growth Factor

Molecular genesis of cutaneous melanoma pathways; rous avian sarcoma homologue (Ras); v-raf murine sarcoma viral oncogene homologue B1 (BRAF); mitogen- activated protein kinase (MEK); extracellular signal-regulated kinase (ERK); microphthalmia transcription factor (Mitf); phosphatidylinositol-3 kinase (PI3K); murine v-akt oncogene homologue (Akt). mammalian target of rapamycin (mTOR); cyclin dependent kinase inhibitor-2A
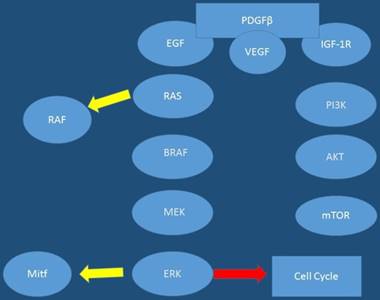
Moreover; in addition to the various mutations that have been highlighted and interact in key component of signaling cascades, different types of RNAs have been shown to be involved in melanoma pathogenesis such as: miRNAs and lncRNAs [48, 49]. It has been observed that various miRNAs have been implicated in the processes of carcinogenesis leading to melanoma. Up-to date data the following miRNAs to be upregulated in melanoma: miR-101, -182, -221, -222, -106-363, -106a, -92, -196, -21, -156, -214, -30b, -30d and -532-5p. Those shown to be downregulated or lost in melanoma are: Let7a and b, miR-31, -125b, -148a, -211, -193b, -196a-1, -196a-2, and -203 [50]. Furthermore, human melanoma cells previously were shown to have higher levels of the long non-coding RNA (lncRNA), SPRINGTLY, compared to normal human melanocytes [48]. In the study by Zhao and co-workers it was observed that stable clones isolated from introduction of SPRINGTLY in human melanocytes led to increased cell proliferation, reduction in apoptosis, invasion, colony formation, and development of a multinucleated dendritic-like phenotype. Moreover; in this study when siRNA was used to knockdown SPRINGTLY, a decrease in cell proliferation, invasion and increase in pro-apoptotic signals was detected [48]. Figures 1,2
Discussion
It has been observed that currently the best treatment for cutaneous early stage melanoma surgical resection with adequate margins is the best choice [51, 52]. In the case of high risk features such as regional lymph node involvement, then adjuvant interferon alpha may be considered [53]. It has been observed that when melanoma patients undergo chemotherapy, there is the possibility that the tumors adapt and become resistant against monotherapies. Therefore, in order to increase the efficacy of therapeutic treatments, combination therapies are commonly administered. Previosly in a preclinical study BRAF inhibitor-resistant mutated BRAF melanoma cells were developed as an experimental model system to mimic BRAF inhibitor-resistant melanoma patients and to study the mechanisms of resistance. In this study these cells were treated with a combination of gamma secretase inhibitor and BRAF inhibitor, a reduction in cell growth and an increase in senescence was observed in these resistant cells. Moreover; when they were weaned off GSI during the study, there was an increase in cell growth [54]. Based on these findings it is suggested that a combination therapy, which inhibits both MAPK and NOTCH pathways, may improve the efficacy of melanoma patients who develop BRAF inhibitor-resistance, however; both inhibitors must be present to sustain the anti-tumor progression response. Furthermore; recently, anti CTLA-4 therapy in adjuvant settings has also shown increased benefit. It is considered that in advanced or metastatic disease, immune checkpoint inhibitors, such as anti PD-1/PDL-1 and CTLA4 should be the standard of care [55]. It is known that many drugs used in the treatment of various types of cancer including melanoma are targeting Met and hepatocyte growth factor (HGF) (or scatter factor (SF) pathways. Additionally, several studies indicated that Met inhibitors should be used in the treatment of cutaneous melanoma in different regimens/combinations [55]. Regarding cutaneous melanoma treatment, multitarget treatment should be considered targeting several molecular pathways at the same time instead of a single agent. These multi-target therapeutic strategies for the management of cutaneous melanoma suggest using combination of HGF/SF-Met inhibitors and chemotherapeutic drugs. Hepatocyte growth factor (HGF) (or scatter factor (SF) pathways -Met inhibitors could be used in combination with other treatment modalities such as; radiotherapy and/or immunotherapy in treatment of cutaneous melanoma. Currently it is suggested that radiotherapy should be considered when the wide excision of the primary site is not possible. Moreover; superficial melanoma, and mucosal melanoma can be treated with local radiotherapy [56, 57]. Previously published retrospective clinical studies and in vitro data regarding the association of radiotherapy with inhibitors of mutant BRAF kinase (vemurafenib) suggest a radiosen-sitization (cell cycle arrest in G1, DNA repair decrease, restore dradiation-induced apoptosis) and an increased radiation-induced toxicity [56, 58]. Radio sensitizers and novel techniques of radiotherapy have improved clinical outcomes in melanoma. However; more clinical trials are needed to clarify indications and therapeutic index. Moreover; understanding the mechanisms of migration, invasion and pre-metastatic formation is crucial on developing novel therapies and using correctly the current ones. Furthermore; current research in different melanoma exosome-specific proteins found in the circulating exosomes of patients have been shown to correlate with prognosis. These proteins can be used in cancer detection and estimation of prognosis. Moreover; advances in genome engineering through CRISPR/Cas9 technology will provide an effective platform on which to perform large-scale screens and identify new synthetic lethal partners. Building upon existing knowledge about gene pair interactions future drugs will be created. Future discoveries will include multimodality treatment. Table 1.
Systemic therapy options for patients with advanced melanoma, according to NCCN guidelines.
| First-Line Agents |
| Immunotherapy |
| A) Anti PD-1 monotherapy - Pembrolizumab _ Nivolumab _ Nivolumab/ipilimumab |
| B) Targeted therapy if BRAF mutated; preferred if clinically needed for early response |
| C) Combination therapy (preferred) - Dabrafenib/trametinib _ Vemurafenib/cobimetinib |
| D) Single-agent therapy: - Vemurafenib _ Dabrafenib _ Clinical trial |
| Second-Line Agents |
| Same as first line, plus: _ High-dose IL-2 _ Biochemotherapy _ Vemurafenib _ Cytotoxic agents_ |
| Imatinib for tumors with activating mutations of c-KIT _ Clinical trials 196 D.G.F. Al-U'datt et al. / Biomedicine & Pharmacotherapy 88 (2017) 194-202 |
Competing Interests
The authors have declared that no competing interest exists.
References
1. Fiessler C, Pfahlberg AB, Keller AK, Radespiel-Troger M, Uter W, Gefeller O. Association between month of birth and melanoma risk: fact or fiction? International journal of epidemiology. 2016 doi:10.1093/ije/dyw226
2. Ziefle S, Egberts F, Heinze S, Volkenandt M, Schmid-Wendtner M, Tilgen W. et al. Health-related quality of life before and during adjuvant interferon-alpha treatment for patients with malignant melanoma (DeCOG-trial). Journal of immunotherapy. 2011;34:403-8 doi:10.1097/CJI.0b013e31821b7a4b
3. Wieder T, Brenner E, Braumuller H, Rocken M. Immunotherapy of melanoma: efficacy and mode of action. Journal der Deutschen Dermatologischen Gesellschaft = Journal of the German Society of Dermatology: JDDG. 2016;14:28-37 doi:10.1111/ddg.12819
4. Brigati C, Noonan DM, Albini A, Benelli R. Tumors and inflammatory infiltrates: friends or foes? Clinical & experimental metastasis. 2002;19:247-58
5. Chattopadhyay S, Mehrotra S, Chhabra A, Hegde U, Mukherji B, Chakraborty NG. Effect of CD4+CD25+ and CD4+CD25- T regulatory cells on the generation of cytolytic T cell response to a self but human tumor-associated epitope in vitro. Journal of immunology. 2006;176:984-90
6. Fidler IJ. Inhibition of pulmonary metastasis by intravenous injection of specifically activated macrophages. Cancer research. 1974;34:1074-8
7. Kozlowska K, Cichorek M. Heterogeneity of peritoneal macrophages in hamsters-bearing transplantable melanomas in relation to their transglutaminase. Folia histochemica et cytobiologica. 1991;29:141-7
8. Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR. et al. Final version of 2009 AJCC melanoma staging and classification. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2009;27:6199-206 doi:10.1200/JCO.2009.23.4799
9. Korn EL, Liu PY, Lee SJ, Chapman JA, Niedzwiecki D, Suman VJ. et al. Meta-analysis of phase II cooperative group trials in metastatic stage IV melanoma to determine progression-free and overall survival benchmarks for future phase II trials. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2008;26:527-34 doi:10.1200/JCO.2007.12.7837
10. Merrill SJ, Subramanian M, Godar DE. Worldwide cutaneous malignant melanoma incidences analyzed by sex, age, and skin type over time (1955-2007): Is HPV infection of androgenic hair follicular melanocytes a risk factor for developing melanoma exclusively in people of European-ancestry? Dermato-endocrinology. 2016;8:e1215391. doi:10.1080/19381980.2016.1215391
11. Ascierto PA, Marincola FM. 2015: The Year of Anti-PD-1/PD-L1s Against Melanoma and Beyond. EBioMedicine. 2015;2:92-3 doi:10.1016/j.ebiom.2015.01.011
12. Ascierto PA, Kirkwood JM, Grob JJ, Simeone E, Grimaldi AM, Maio M. et al. The role of BRAF V600 mutation in melanoma. Journal of translational medicine. 2012;10:85. doi:10.1186/1479-5876-10-85
13. Konieczkowski DJ, Johannessen CM, Abudayyeh O, Kim JW, Cooper ZA, Piris A. et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer discovery. 2014;4:816-27 doi:10.1158/2159-8290.CD-13-0424
14. Muller J, Krijgsman O, Tsoi J, Robert L, Hugo W, Song C. et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nature communications. 2014;5:5712. doi:10.1038/ncomms6712
15. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S. et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165:35-44 doi:10.1016/j.cell.2016.02.065
16. Little AS, Smith PD, Cook SJ. Mechanisms of acquired resistance to ERK1/2 pathway inhibitors. Oncogene. 2013;32:1207-15 doi:10.1038/onc.2012.160
17. Wang S, Fan W, Wan B, Tu M, Jin F, Liu F. et al. Characterization of long noncoding RNA and messenger RNA signatures in melanoma tumorigenesis and metastasis. PloS one. 2017;12:e0172498. doi:10.1371/journal.pone.0172498
18. Pasquali S, Chiarion-Sileni V, Rossi CR, Mocellin S. Immune checkpoint inhibitors and targeted therapies for metastatic melanoma: A network meta-analysis. Cancer treatment reviews. 2017;54:34-42 doi:10.1016/j.ctrv.2017.01.006
19. Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2006;24:4340-6 doi:10.1200/JCO.2006.06.2984
20. Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H. et al. Distinct sets of genetic alterations in melanoma. The New England journal of medicine. 2005;353:2135-47 doi:10.1056/NEJMoa050092
21. Bastian BC, Kashani-Sabet M, Hamm H, Godfrey T, Moore DH 2nd, Brocker EB. et al. Gene amplifications characterize acral melanoma and permit the detection of occult tumor cells in the surrounding skin. Cancer research. 2000;60:1968-73
22. Bennett DC. Human melanocyte senescence and melanoma susceptibility genes. Oncogene. 2003;22:3063-9 doi:10.1038/sj.onc.1206446
23. Michaloglou C, Vredeveld LC, Mooi WJ, Peeper DS. BRAF(E600) in benign and malignant human tumours. Oncogene. 2008;27:877-95 doi:10.1038/sj.onc.1210704
24. Miller AJ, Mihm MC Jr. Melanoma. The New England journal of medicine. 2006;355:51-65 doi:10.1056/NEJMra052166
25. Bishop DT, Demenais F, Goldstein AM, Bergman W, Bishop JN, Bressac-de Paillerets B. et al. Geographical variation in the penetrance of CDKN2A mutations for melanoma. Journal of the National Cancer Institute. 2002;94:894-903
26. Grossman D, Leffell DJ. The molecular basis of nonmelanoma skin cancer: new understanding. Archives of dermatology. 1997;133:1263-70
27. Meyskens FL Jr, Farmer PJ, Anton-Culver H. Etiologic pathogenesis of melanoma: a unifying hypothesis for the missing attributable risk. Clinical cancer research: an official journal of the American Association for Cancer Research. 2004;10:2581-3
28. Brown VL, Harwood CA, Crook T, Cronin JG, Kelsell DP, Proby CM. p16INK4a and p14ARF tumor suppressor genes are commonly inactivated in cutaneous squamous cell carcinoma. The Journal of investigative dermatology. 2004;122:1284-92 doi:10.1111/j.0022-202X.2004.22501.x
29. Ohtani N, Yamakoshi K, Takahashi A, Hara E. The p16INK4a-RB pathway: molecular link between cellular senescence and tumor suppression. The journal of medical investigation: JMI. 2004;51:146-53
30. Thompson JF, Scolyer RA, Kefford RF. Cutaneous melanoma. Lancet. 2005;365:687-701 doi:10.1016/S0140-6736(05)17951-3
31. Kefford RF, Mann GJ. Is there a role for genetic testing in patients with melanoma? Current opinion in oncology. 2003;15:157-61
32. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S. et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949-54 doi:10.1038/nature00766
33. Pollock PM, Harper UL, Hansen KS, Yudt LM, Stark M, Robbins CM. et al. High frequency of BRAF mutations in nevi. Nature genetics. 2003;33:19-20 doi:10.1038/ng1054
34. Wangari-Talbot J, Chen S. Genetics of melanoma. Frontiers in genetics. 2012;3:330. doi:10.3389/fgene.2012.00330
35. Jakob JA, Bassett RL Jr, Ng CS, Curry JL, Joseph RW, Alvarado GC. et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer. 2012;118:4014-23 doi:10.1002/cncr.26724
36. Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O'Brien JM. et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457:599-602 doi:10.1038/nature07586
37. Chen S, Zhu H, Wetzel WJ, Philbert MA. Spontaneous melanocytosis in transgenic mice. The Journal of investigative dermatology. 1996;106:1145-51
38. Zhu H, Reuhl K, Zhang X, Botha R, Ryan K, Wei J. et al. Development of heritable melanoma in transgenic mice. The Journal of investigative dermatology. 1998;110:247-52 doi:10.1046/j.1523-1747.1998.00133.x
39. Namkoong J, Shin SS, Lee HJ, Marin YE, Wall BA, Goydos JS. et al. Metabotropic glutamate receptor 1 and glutamate signaling in human melanoma. Cancer research. 2007;67:2298-305 doi:10.1158/0008-5472.CAN-06-3665
40. Teh JL, Chen S. Glutamatergic signaling in cellular transformation. Pigment cell & melanoma research. 2012;25:331-42 doi:10.1111/j.1755-148X.2012.00983.x
41. Gilman AG. G proteins: transducers of receptor-generated signals. Annual review of biochemistry. 1987;56:615-49 doi:10.1146/annurev.bi.56.070187.003151
42. Yu LJ, Wall BA, Wangari-Talbot J, Chen S. Metabotropic glutamate receptors in cancer. Neuropharmacology. 2016 doi:10.1016/j.neuropharm.2016.02.011
43. Wellbrock C, Ogilvie L, Hedley D, Karasarides M, Martin J, Niculescu-Duvaz D. et al. V599EB-RAF is an oncogene in melanocytes. Cancer research. 2004;64:2338-42
44. Shin SS, Namkoong J, Wall BA, Gleason R, Lee HJ, Chen S. Oncogenic activities of metabotropic glutamate receptor 1 (Grm1) in melanocyte transformation. Pigment cell & melanoma research. 2008;21:368-78 doi:10.1111/j.1755-148X.2008.00452.x
45. Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE Jr. et al. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nature genetics. 2009;41:544-52 doi:10.1038/ng.356
46. Jimenez-Garcia L, Herranz S, Higueras MA, Luque A, Hortelano S. Tumor suppressor ARF regulates tissue microenvironment and tumor growth through modulation of macrophage polarization. Oncotarget. 2016;7:66835-50 doi:10.18632/oncotarget.11652
47. Wilkins DK, Nathan PD. Therapeutic opportunities in noncutaneous melanoma. Therapeutic advances in medical oncology. 2009;1:29-36 doi:10.1177/1758834009337664
48. Zhao W, Mazar J, Lee B, Sawada J, Li JL, Shelley J. et al. The Long Noncoding RNA SPRIGHTLY Regulates Cell Proliferation in Primary Human Melanocytes. The Journal of investigative dermatology. 2016;136:819-28 doi:10.1016/j.jid.2016.01.018
49. Gasque Schoof CR, Izzotti A, Jasiulionis MG, Vasques Ldos R. The Roles of miR-26, miR-29, and miR-203 in the Silencing of the Epigenetic Machinery during Melanocyte Transformation. BioMed research international. 2015;2015:634749. doi:10.1155/2015/634749
50. Gajos-Michniewicz A, Duechler M, Czyz M. MiRNA in melanoma-derived exosomes. Cancer letters. 2014;347:29-37 doi:10.1016/j.canlet.2014.02.004
51. Dummer R, Hauschild A, Guggenheim M, Jost L, Pentheroudakis G, Group EGW. Melanoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of oncology: official journal of the European Society for Medical Oncology. 2010;21(Suppl 5):v194-7 doi:10.1093/annonc/mdq188
52. Dummer R, Siano M, Hunger RE, Lindenblatt N, Braun R, Michielin O. et al. The updated Swiss guidelines 2016 for the treatment and follow-up of cutaneous melanoma. Swiss medical weekly. 2016;146:w14279. doi:10.4414/smw.2016.14279
53. Mocellin S, Pasquali S, Rossi CR, Nitti D. Interferon alpha adjuvant therapy in patients with high-risk melanoma: a systematic review and meta-analysis. Journal of the National Cancer Institute. 2010;102:493-501 doi:10.1093/jnci/djq009
54. Zhu G, Yi X, Haferkamp S, Hesbacher S, Li C, Goebeler M. et al. Combination with gamma-secretase inhibitor prolongs treatment efficacy of BRAF inhibitor in BRAF-mutated melanoma cells. Cancer letters. 2016;376:43-52 doi:10.1016/j.canlet.2016.03.028
55. Al-U'datt D G, Al-Husein BA, Qasaimeh GR. A mini-review of c-Met as a potential therapeutic target in melanoma. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2017;88:194-202 doi:10.1016/j.biopha.2017.01.045
56. Fort M, Guet S, Husheng S, Calitchi E, Belkacemi Y. et al. Erratum to "Role of radiation therapy in melanomas: Systematic review and best practice in 2015" [Crit. Rev Oncol/Hematol 99 (2016) 362-375]. Critical reviews in oncology/hematology. 2016;102:144. doi:10.1016/j.critrevonc.2016.04.008
57. Mahadevan A, Patel VL, Dagoglu N. Radiation Therapy in the Management of Malignant Melanoma. Oncology. 2015;29:743-51
58. Sambade MJ, Peters EC, Thomas NE, Kaufmann WK, Kimple RJ, Shields JM. Melanoma cells show a heterogeneous range of sensitivity to ionizing radiation and are radiosensitized by inhibition of B-RAF with PLX-4032. Radiotherapy and oncology: journal of the European Society for Therapeutic Radiology and Oncology. 2011;98:394-9 doi:10.1016/j.radonc.2010.12.017
Author contact
![]() Corresponding author: Paul Zarogoulidis, M.D, Ph. D, Pulmonary Department-Oncology Unit, “G. Papanikolaou” General Hospital, Aristotle University of Thessaloniki, Thessaloniki, Greece Fax: 00302310992424 Mobile: 00306977271974 E-mail: pzarogcom
Corresponding author: Paul Zarogoulidis, M.D, Ph. D, Pulmonary Department-Oncology Unit, “G. Papanikolaou” General Hospital, Aristotle University of Thessaloniki, Thessaloniki, Greece Fax: 00302310992424 Mobile: 00306977271974 E-mail: pzarogcom