J Biomed 2018; 3:50-59. doi:10.7150/jbm.27148 This volume Cite
Research Paper
Poly(lactic-co-glycolic acid) based double emulsion nanoparticle as a carrier system to deliver glutathione sublingually
Guanyu Chen, Jingyuan Wen ![]()
School of Pharmacy, Faculty of Medical and Health Science, University of Auckland, New Zealand
Received 2018-5-8; Accepted 2018-6-9; Published 2018-6-24
Abstract

The sublingual delivery of drugs represents a continuing challenge, as well as an opportunity. In this study, a poly(lactic-co-glycolic acid) (PLGA) based Water-in-Oil-in-Water (w/o/w) double emulsion of nanoparticles was developed as potential platform technology, to generate sustained drug release profile and maximize the bioavailability of the glutathione (GSH). Two-level full factorial design was carried out based on different types and concentrations of the stabilizers used, and on varying sonication time. The optimal formulations were predicted with particle size of 232.57 ± 20.56 nm, zeta-potential of -12.33 ± 0.20 mV and entrapment efficiency of 77.04 ± 1.50%. Spherical particle morphology and uniform size was observed by scanning electron microscopy. Sustained release profile was achieved and the release kinetics was illustrated as a Korsmeyer-Peppas kinetic model. Moreover, in ex-vivo permeation studies over porcine mucosal epithelial membrane, approximately 2-fold increase in the transmucosal permeation of GSH was achieved when PVA and carbopol were incorporated with the drug delivery system, resulting in 21.9 ± 1.2% drug permeation and a permeation coefficient of (4.96 ± 0.36) × 10-6 cm∙sec-1. The developed PLGA nanoparticulate delivery system was able to elevate the retention time and resist saliva dissolution, providing a sustained drug release profile and relatively high transmucosal permeation of GSH.
Keywords: Sublingual delivery, PLGA Nanoparticles, Factorial design, Glutathione, Sustained release, Transmucosal permeation
Introduction
GSH is one of the most essential and powerful antioxidants in the human body system. It helps recycle vitamin C and E, while maintaining its antioxidant activities. It may well be the most important antioxidant that the cells of our body produce, helping to neutralize reactive oxygen compounds and free radicals.1 Unfortunately, increasing the body's levels of GSH is not always easy, as much of the substance is lost in the digestive system, and the small amount that does get through is usually insufficient to be of any real benefit. Moreover, because it has a very short plasma half-life of approximate 2.5 min.2 Therefore, a novel drug delivery system which is able to achieve sustained drug release and greater bioavailability is desirable.
Sublingual drug delivery involves delivering the drug to the sublingual region of the oral cavity, through the mucosal membrane into the systemic circulation. The application of drugs to the surface of mucosal tissue is a convenient and painless method of administration, bypassing the hepatic first-pass effect and avoiding enzymatic degradation in the gastrointestinal tract. It is necessary for the drugs to be retained for a sufficiently long period of time, to achieve the desired therapeutic effect.3 However, disadvantages are also involved in this delivery route. The small available surface region of administration site is one disadvantage and the fact that saliva may wash off the dosage is a main concern with regard to retention of the drug components.4 Moreover, there are the physical barriers and the biochemical barriers to sublingual delivery. The physical barriers are include the saliva and mucus layer, mucosal epithelium and the basement membrane, while with biochemical barriers, the drug candidates can be degraded by the metabolizing enzymes from the sublingual region.5
Nanoparticles (nanospheres) for sublingual drug delivery can be defined as submicron (1 to 1000 nm) colloidal particles from natural or artificial polymers, in which the drug molecule is adsorbed, dissolved or dispersed throughout the matrix.6 The advantages of using nanoparticles as drug carriers for sublingual delivery include: high carrier capacity; the capability of both hydrophobic and hydrophilic drugs to be encapsulated allowing controlled drug release; prevention of enzymatic degradation; versatility of the nanoparticles and their availability in a wide range of modifiable structures, which help in targeting specific sites of action; generation of good stability meaning longer shelf life.7 Poly(lactic-co-glycolic acid) (PLGA) is a copolymer which is used in a host of Food and Drug Administration (FDA) approved therapeutic devices. Owing to its biodegradability and biocompatibility, it does not lead to polymer accumulation in the body as it has a natural degradability to non-toxic metabolites that can then be excreted from the body.8 Indeed PLGA particles are extensively investigated for drug delivery, but there are still improvements needed in the existing methods to overcome the difficulties in terms of reproducibility, size, and shape. The size and shape of the colloidal particles are influenced by the stabilizer and the solvent used.9 Therefore in this study, different types and concentrations of stabilizer were investigated such as polyvinyl alcohol BF17W (PVA), carbopol, methyl cellulose (MC), hydroxyl propyl pyrolidine (HPMC) and poloxamer. It is suggested that a fraction of PVA always remains associated with the nanoparticles despite repeated washings because PVA forms an interconnected network with the polymer at the interface.10 Moreover, the stability and biological activity of the nanoparticles have been major concerns due to the involvement of organic solvents during the preparation process.11
A strategy for evaluation and optimization of the parameters of delivery systems in an efficient approach is necessary. Two-level full factorial design was applied in this study to optimize the pharmaceutical process to yield a maximum or minimum response based on the relationship between dependent variables (responses) and controllable (independent) variables. Response Surface Methodology (RSM) is a collection of mathematical and statistical techniques that can be used to develop, improve or optimize products. RSM typically is used for the modelling and analysis of problems in which a response of interest is affected by several variables with the objective of optimizing this response.
It appears that using GSH-loaded polymeric nanoparticles for sublingual delivery has not yet been reported in the literature. The aim of this project is to develop a GSH-loaded PLGA based double emulsion of nanoparticles as a carrier delivery system via sublingual administration, to achieve improved bioavailability in a controlled release manner. The present work deals with the fabrication of PLGA nanoparticles by solvent evaporation method and optimal formulations were predicted by factorial design method based on particle size and EE results, in-vitro drug release profile and ex-vivo drug permeation studies were investigated. The results indicated that the developed optimal PLGA nanoparticulate delivery system can provide a promising formulation platform from which to develop commercial products of GSH for sublingual drug delivery.
Materials and Methods
Materials
PLGA was supplied by Purac Biochem, USA. PVA was purchased from Ajax Finechem, Australia. Carbopol 971NF was obtained from Noveon, USA. MC was supplied by ICN Biomedicals, USA. Chitosan was obtained from Fluka, Israel. Poxolamer 188, HPMC and GSH were purchased from Sigma-Aldrich, USA. Ethyl acetate, acetic acid, Milli-Q water were laboratory analytical grade.
Preparation of nanoparticles
PLGA dissolved in 3 mL of ethyl acetate, and 0.6 mL of 1% w/v drug solution was added dropwise into PLGA solution forming a w/o emulsion and was sonicated at an output of 20 W for 60 seconds by ultrasonicator (Model UP 200S, Hielscher, Germany). This was then poured into a solution of PVA, and sonicated at an output of 20 W for the required time to form a w/o/w double emulsion. Solvent was evaporated using a rotator evaporator (V-850 model, Buchi®, USA) after which the resulting nanosuspension was centrifuged with relative centrifugal force of 173210 g for 40 mins to harvest the nanoparticles by using Ultracentrifuge (Wx Ultra 80 model, Thermo Scientific®, USA), washed thoroughly by 5% w/v sucrose solution twice and freeze-dried for 24 hours at -50 ºC (Model Freezone 6, Labconco, USA).
Characterization
Factorial design
A two-level full factorial design method (Design-Expert®, version 7.0, State-Ease inc, Minneapolis, MN, USA) was applied to predict the optimal formulation. The three independent parameters were different stabilizers, different concentration of the stabilizers used, and different sonication times applied during the preparation procedure. The normalized factor levels of independent parameters are given in Table 1. Particle size and EE were evaluated as dependent variables, which were also defined as response. The total number of formulations was automatically generated by the software subjected to the level of factors in Table 1. Particle size and EE of the formulations were determined. Then the software could predict the optimal formulation according to the desirability of smaller particle size and higher EE.12
The independent and dependent variables in two level full factorial design.12
| Level of factors in design | |||
|---|---|---|---|
| Low (-) | Moderate (0) | High (+) | |
| PVA | 0.000% w/v | 0.500% w/v | 1.000% w/v |
| Carbopol | 0.006% w/v | 0.012% w/v | 0.024% w/v |
| MC | 0.038% w/v | 0.076% w/v | 0.152% w/v |
| HPMC | 0.038% w/v | 0.076% w/v | 0.152% w/v |
| Poloxamer | 1.100% w/v | 2.199% w/v | 4.398% w/v |
| Sonication time | 180 sec | 300 sec | 480 sec |
Particle size, size distribution and zeta potential
The particle size, size distribution and zeta potential of nanoparticle were measured by Malvern Zetasizer (Zetasizer Nano ZS, Malvern, UK). Nanosuspension sample before freeze-dried was diluted 20 times with milli-Q water prior to the measurements. The Average diameter of the particle (Z ave) and the zeta potential (mV) was determined.
Entrapment efficiency
The nanosuspension sample was centrifuged, 1 mL of the supernatant containing unentrapped GSH was filtered through a 0.22 µm MS® PES syringe filter and analysed by HPLC. The EE was calculated as Equation 1:
Equation 1
The factorial design software predicted the optimal formulations with smaller particle size and greater EE. The formulations were used to further conduct in-vitro drug release and ex-vivo drug permeation studies.
In-vitro drug release studies
The in-vitro drug release was carried out using Franz diffusion cells (VTC 200, Logan, USA) with cellulose membrane (MW range from 8000 Da to 14000 Da) sandwiched between donor and receptor chambers. Sodium dihydrogen phosphate monohydrate buffer (0.1 M, pH 6.5) containing 13.8 g/L of sodium dihydrogen phosphate monohydrate was used as the medium, while the temperature was maintained at 37 ± 1 °C. GSH (1% w/v), and drug loaded nanoparticles with equivalent GSH amount, were added to the donor chamber. An aliquot sample of 0.5 mL was withdrawn at different time intervals over 12 hours, and replaced by 0.5 mL of fresh medium. All the samples were filtered by 0.22 µm MS® PES syringe filters prior to HPLC determination.13 All experiments were conducted with triplicates, and results are given as mean ± SD.
In-vitro release kinetic modelling
Model dependent methods are based on different mathematical functions, which describe the dissolution profile (Table 2). Once a suitable function has been selected, the dissolution profiles are evaluated depending on the derived model parameters. In order to investigate the mode of release from the optimized formulations, the previous in-vitro release data were analysed using the following mathematical kinetic models, in order to understand the drug release mechanisms and the kinetics.14
Mathematical kinetic models for drug release studies.14,15
| Kinetic models | Equations | Abbreviations |
|---|---|---|
| Zero order | C = k0t | Where, K0 is zero-order rate constant expressed in units of concentration versus time and t is the time. |
| First order | LogC = LogCo - k.t/2.303 | C0 is the initial concentration of drug and K is first order constant and t is the time. |
| Higuchi model | Q = Kt1/ 2 | Where, K is the constant reflecting the design variables of the system. Hence drug release rate is proportional to the reciprocal of the square root of time. |
| Korsmeyer-Peppas models |  = K t n = K t n | Where  is fraction of drug released at time t, k is the rate constant and n is the release exponent indicative of the mechanism of release. is fraction of drug released at time t, k is the rate constant and n is the release exponent indicative of the mechanism of release. |
Ex-vivo drug permeation studies
The Franz diffusion cells were also used in this study. Permeability was measured with porcine mucosal epithelial membrane soaked in 0.1 M sodium dihydrogen phosphate monohydrate buffer (pH 6.5) overnight, and sandwiched in between the donor and receptor chambers. GSH (1% w/v), and drug loaded nanoparticles that were equivalent to 1% w/v GSH were added onto the mucosal surface in the donor compartment. An aliquot sample of 0.5 mL was withdrawn at various time intervals from the receptor chamber and filtered with 0.22 µm MS® PES syringe filters. The same volume of medium was replaced in the receptor chamber after each sampling.16 The concentration of gemcitabine was then determined by HPLC. All experiments were conducted with triplicates, and results are given as mean ± SD.
Scanning Electronic Microscopy (SEM) morphology studies
Small amounts of freeze-dried nanoparticles samples were placed on a grid and dried at room temperature overnight and sputter-coated with gold and palladium. They were then observed under a condition of high vacuum and temperature of less than -120 ºC using SEM (XL30S FEG model, Philips. USA) at 25 kV.
Statistical analysis
All data reported are means of triplicates, with standard deviation as the errors. Additionally, differences in Papp between different formulation groups were evaluated using one-way ANOVA followed by Tukey's test. Probability estimates of P < 0.05 were considered to be significant. All analyses were carried out using MINITAB (version 17).
Results and Discussions
Particle size and size distribution
Particle size is a key factor influencing the rate and extent of permeation across the cell membrane as well as the stability of the formulation. Large particle size distribution might lead to aggregation thus slow drug release rate and reduction in permeation as a result.17 The results showed that particle sizes ranging from 100 to 500 nm were measured, a size beneficial for drug permeation over oral mucosa tissue.18 PVA carbopol group showed the relatively smaller particle size compared to other groups. Additionally, particle size of 232.57 ± 10.56 nm with PdI of 0.278 was obtained with carbopol combining with PVA and used as stabilizer, which appeared smallest among the formulation. This can be explained by the fact that carbopol itself is a good stabilizer.19 Moreover, The PVA is known to form layers of aggregates around the surface of nanoparticles contributing towards the hydrodynamic diameter of nanoparticles.20 After the removal of organic solvent, more PVA stabilizer molecules can be physically incorporated onto the surface of nanoparticles and then a large number of hydroxyl groups extending into the continuous phase could be hydrated, forming a surface layer that will hinder nanoparticle aggregation.21
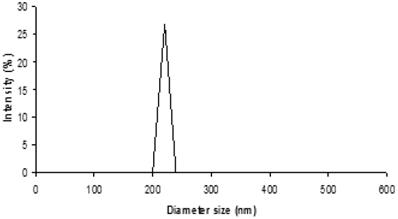
From Fig. 1, the size distribution of one of the optimal formulation has shown an intensity peak with relatively high percentage intensity indicating the good uniformity of the particles of this formulation and low particle aggregation and drug leakage.22 In addition, the polydispersity index (PdI) describes variation in sizes. The higher the PdI value, means the wider the particle size distribution and vice versa.23 For this formulation, the low PdI value indicates the promising uniformity of the particles.
Size distribution output of the formulation 0.2% w/v PVA and 0.03% w/v carbopol with 480 sec sonication time. (PdI: 0.278).
Morphology studies
Scanning electronic microscopy showed that isolated PLGA nanoparticles were uniform, spherical and well defined in shape with smooth surface (Fig. 2). When PVA was alone used in the preparation the particle size was about 250 nm. It showed that when other stabilizing polymers were combined to use, SEM analysis showed larger particle size of over 300 nm. PVA and carbopol formulation appeared to have smallest size distribution, indicating better size uniformity of the formulation. However, some of the particles had aggregated due to the high energy centrifugation that forces the particle back together to form aggregates.24
Zeta potential
The zeta potential of all the formulations appeared to have negative values that were within a range from -0.10 mV to -12.5 mV. The negativity ranking of zeta potential of the four groups is PVA and carbopol > PVA and poloxamer > PVA and HPMC > PVA and MC. From the results, polymers carbopol and poloxamer were found to have a significant influence on the zeta potential value of the PLGA nanoparticles. The optimal formulation of PVA and carbopol had a relatively greater negative value of zeta potential of -12.33 ± 0.10 mV, which indicated the more stable of the formulations.25 The zeta potential value is an important particle characteristic as it can influence both particle stability as well as particle mucoadhesion. This is due to the fact that zeta potential represents the stability of the colloidal dispersion and the extent of repulsion between nanoparticles, thus there is less likelihood of aggregation due to the presence of the same charge layer which will repel the nanoparticles away from each other.25 In theory, more pronounced zeta potential values, being positive (≥ 30 mV) or negative (≤ -30 mV), tend to stabilize particle suspensions.26 The optimal formulation of PVA and carbopol had a relatively larger negative value of zeta potential which is preferred. This may be due to the fact that PVA and carbopol contain high proportions of hydroxyl and carboxyl groups respectively, therefore these create a negative shell layer surrounding the nanoparticles.27
The SEM micrograph of nanoparticles of PVA and carbopol formulation (left, magnification: 25000×), and the nanoparticles of PVA and poloxamer formulation (right, magnification: 50000×).

Entrapment efficiency
The range of EE of the formulations was from 66% to 79%. And the optimal formulation had an EE of 77.04 ± 0.50%. There was a trend of an increase of EE when the PVA concentration increases. A possible explanation for this phenomenon is that an increase in PVA concentration may increase the viscosity of the external water phase. This in turn elevates the difficulty for diffusion of the drug into the external water phase, which in turn leads to increase drug retention in the particle and a higher EE.28 In other words, the presence of increasing PVA concentration might serve as a barrier to prevent the diffusion of the drug out of the polymeric shell, resulting in the gradual increase of EE when the PVA concentration is elevated.29 Another phenomenon was an increase in EE when the 2nd sonication duration was reduced from 180 sec to 480 sec for 2nd sonication duration time when EE was reduced about 5%. This could be explained by the increased sonication time leading to breakage of the particles, and resulting in leakage of drug,30 thereby a lower EE was obtained. Moreover, the 30% unentrapped drug that was determined might have been due to the fact that the precipitation of PLGA was not fast enough for the drug to diffuse into the outer aqueous phase during the fabrication process. It is known that the amount of drug entrapped within the particles is dependent on the speed of polymer precipitation from the organic phase.28 GSH is very soluble in the outer aqueous phase. This property coupled with the concentration gradient between the inner and outer aqueous phases drives GSH to diffuse into the outer aqueous medium via the organic layer.31 Additionally, subjecting the particles for centrifugation to precipitate the particles from the supernatant could also cause the release of some GSH during the process.32 Furthermore, the lack of sufficient time to allow PLGA to undergo polymerization could account for less drug entrapment in the PVA stabilized loaded particles.31
Prediction of optimal formulations by factorial design
All the data generated from particle size and EE of the formulations were entered into the factorial design program and the optimal formulation was predicted based on the desired response of smallest particle size and highest EE. The two resultant optimal formulations are (i) 0.20% w/v PVA and 0.03% w/v carbopol with 480 sec 2nd sonication time (Desirability: 0.917), and (ii) 2.00% w/v PVA and 1.10% w/v poloxamer with 180 sec 2nd sonication time (Desirability: 0.471). The statistical analysis of variance by ANOVA for selected factorial model was evaluated. All the formulations had a p value 'Prob > F' of between 0.010 to 0.041. Values of 'Prob > F' less than 0.050 indicate model terms are significant. 'Adeq Precision' values of formulations were between 7 to 10. 'Adeq Precision' measures the signal to noise ratio. A ratio greater than 4 is desirable, thus all the formulations indicated an adequate signal, and this model can be used to navigate the design space.12
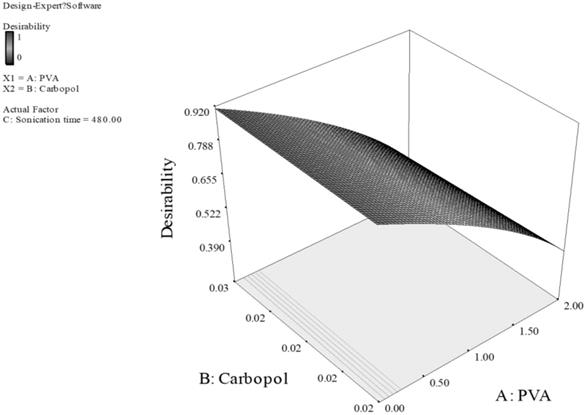
The data was fit to a quadratic model of which the parameters were those of different types of the stabilizer combined, different concentrations used and different 2nd sonication times. The optimal formulation was automated, and a graphical representation of the measured points and the response surface is given in Fig. 3 and Fig. 4.
Response surface of PLGA nanoparticles prepared with PVA and carbopol as stabilizers. (Desirability: 0.917)
Response surface of PLGA nanoparticles prepared with PVA and poloxamer as stabilizers. (Desirability: 0.471)
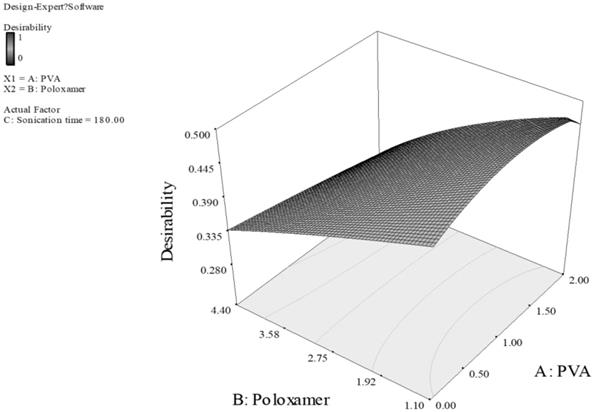
For Fig. 3, the smaller particles and higher EE are obtained at higher level of carbopol and lower level of PVA. This can be explained by the fact that carbopol itself is a good stabilizer for the PLGA emulsion formed during the preparation of the particles.33 With high concentration of both PVA and carbopol, the viscosity of the outer stabilizing aqueous phase was very high. Consequently, at a constant energy input from the ultrasonic probe during the 2nd sonication process, larger emulsion droplets were formed in the w/o/w emulsion, thus larger particles were obtained.33,34 Additionally, sonication time also had a dominant effect on the particle size and EE, the longer sonication time of 480 Sec was predicted for optimal formulation, as the longer sonication time leads to reduction of particle aggregation, thus reducing the particle size. However, the promising stabilizing effect of carbopol resulted in a higher EE. For Fig. 4, concentration of PVA had a dominant effect on particle size and EE while concentration of poloxamer and sonication time showed a minor effect. Poloxamer, in this case was shown to be less favorable than PVA in stabilizing PLGA emulsion, thus the higher PVA concentration generated smaller particle size and greater EE than having greater concentration of poloxamer or both of poloxamer and PVA. Thus, we can conclude that the effect of the concentration of poloxamer is of a much smaller extent on particle size and EE. Consequently, minimizing the particle size and maximizing the EE can be obtained by having higher PVA concentration of 2% w/v, and lower poloxamer concentration of 1.1% w/v.
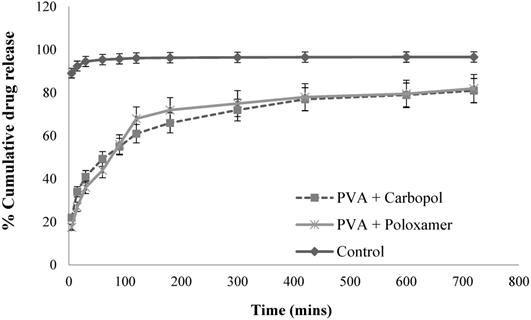
In-vitro release studies of GSH solution and optimal GSH loaded PLGA nanoparticles. (Mean ± SD, n=3).
In-vitro drug release studies
From Fig. 5, for the plain drug solution (the control), over 92.4% of drug was obtained in receptor chamber over 15 mins. For the optimal GSH loaded nanoparticle formulations, they displayed a sustain release profiles. The formulations produced a biphasic drug release profile with the occurrence of burst phase of approximate 20% of drug released within 15 mins, this was caused by the detachment of the GSH adsorbed on the surface of nanoparticles and/or to the release of drug encapsulated near to nanoparticle surface.35 Another explanation of burst release of the formulations was that of the particles having a more porous surface as the external volume was increased. Subsequently, the encapsulated GSH maintained sustained release until approximately 80% of the loaded drug amount was determined within 12 hours. This sustained and prolonged drug release was due to diffusion coupled with erosion. The release process was controlled by more than one process which is discussed in the release kinetic modelling section. The drug loaded nanoparticles did not achieve in 100% release over 12 hours, this may have been due to the presence of the oily phase within the double emulsion nanoparticles, whereby it may have failed to degrade in the medium during the in-vitro drug release study, with the result that some of the drug encapsulated within the nanoparticles was not released.36 In addition, during the preparation of nanoparticle process, the repeated washing by 10% v/v sucrose solution might also cause drug loss to some extent.
In-vitro release kinetic modelling
The modelled kinetic parameters are reported in Table 3. The release data is subjected to goodness of fit test (r2) by linear regression analysis according to the selected release kinetics models.
Release kinetic parameters of the two optimal formulations.
| Zero order | First order | Higuchi model | Korsmeyer-Peppas model | |
|---|---|---|---|---|
| r2 | r2 | r2 | r2 | |
| PVA + Carbopol | 0.5839 | 0.6937 | 0.7849 | 0.9854 |
| PVA + Poloxamer | 0.5347 | 0.6125 | 0.7427 | 0.9824 |
To study the zero order release kinetics, data obtained from in vitro drug release studies were plotted as % cumulative amount of drug released versus time, and this model describes the systems where the drug release is independent of its concentration. Under the zero order kinetic, the drug diffuses out in a controlled manner from the nanoparticles. It was plotted by the cumulative amount of GSH release vs time for zero order kinetics.14 The r2 values for the zero order kinetics are of 0.5839 and 0.5347 for PVA and carbopol and PVA and poloxamer nanoparticles respectively, which are relatively small. This suggests the drug release does not comply with zero order. The first order drug release describes the release from the delivery systems where the rate of release is concentration dependent. This relationship can be used to describe the drug dissolution in pharmaceutical dosage forms such as those containing water-soluble drugs in porous matrices.14 The first order release profile is achieved by plotting the logarithm of cumulative percentage drug remaining vs time. This model shows a relatively low r2 of 0.6937 and 0.6125 for the two formulations, indicating that GSH release occurs independently of the amount of drug remaining in the carrier.14 Higuchi model describes the release of drug from an insoluble matrix, taking into account the volume of the dosage form accessible to the dissolution media changes that occur with time.15 This model can be analysed by plotting the amount of drug released vs square root of time, the process is based on Fickian diffusion. It is characteristic for nanoparticles where drug release is governed by pure diffusion. Penetration of the media into the dosage form is dependent on the matrix porosity and polymer relaxation.37 The r2 values are of 0.7849 and 0.7427 for the two formulations, which are relatively low for fitting into the Higuchi model, indicating that the release of drug from matrix was not significantly driven by Fickian diffusion.37 A Korsmeyer-Peppas model can be obtained by plotting log cumulative percentage of drug released vs log time for the drug release. From the results, GSH release from the two formulations following the Korsmeyer-Peppas model and the goodness fit test r2 are of 0.9854 and 0.9824 for the two formulations. The diffusional release exponent (n) in the model gives an indication of the diffusion mechanism (Table 4). The release exponent n is 0.4605 and 0.4992 for the two formulations, which indicates that the drug release is governed by diffusion through the nanoparticles matrix as well as matrix erosion, so called anomalous diffusion. This anomalous diffusion is evidence that GSH released from the optimized nanoparticles is controlled by more than one process.36, 38
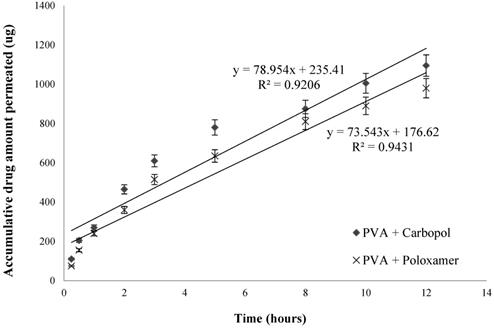
Accumulative GSH amount permeated over porcine mucosal epithelial membrane (n=3, mean ± SD).
Overall in-vitro release data indicates the two optimized formulations are capable of sustained GSH release rate over time. And the drug release kinetics indicate that the drug release is best explained by Korsmeyer-Peppas model, which is indicative of an anomalous diffusion mechanism or diffusion coupled with erosion, confirming that the GSH release from these nanoparticles is controlled by more than one process.
Interpretation of diffusional release mechanisms from nanoparticles.
| Release exponent (n) | Drug transport mechanism | Rate as a function of time |
|---|---|---|
| n = 0.5 | Fickian diffusion | t -0.5 |
| 0.45 < n < 0.89 | Non-Fickian transport | t n-1 |
| 0.89 | Case II (relaxational) transport | Zero order release |
| n > 0.89 | Super case II transport | t n-1 |
Ex-vivo drug permeation studies
From Fig. 6, it showed the transmucosal permeability of GSH released from the nanoparticles of the two selected formulations had a minor variation, with a maximum of 21.9 ± 1.2 % of drug permeated across mucosal epithelial membrane for PVA and carbopol formulation, and 19.6 ± 1.1 % for PVA and poloxamer formulation over 12 hours. The rate of drug permeation was steady, with a faster drug permeation rate for the first 3 hours and subsequently a more gradual permeation profile was observed.
To allow for comparison the permeability of the two optimal formulations, permeation coefficients (Papp) were determined using the following equation 2:
Equation 2
Where, Xr is the accumulative amount of GSH (µg) in receptor chamber, A is the surface area of the mucosal tissue (cm2), and C0 is the initial concentration of GSH in donor chamber (µg∙ mL-1).
The value of dXr/dt is 78.954 and 73.543 for PVA with carbopol, and PVA with poloxamer formulations, respectively (Fig. 6). The surface area A is 1.77 cm2 and C0 is 2500 μg/mL. Therefore, the average value of Papp of PVA with carbopol, and PVA with poloxamer formulations are of (4.96 ± 0.36) × 10-6 cm∙sec-1 and (4.62 ± 0.27) × 10-6 cm∙sec-1, respectively. Both optimized formulations significantly increased the permeability of GSH across the porcine mucosal epithelial membrane compared to that of pure GSH solution, with PVA and carbopol enhancing the permeability of GSH is slightly greater than PVA and poloxamer, as illustrated by their Papp values.
From ex-vivo permeation studies, it was demonstrated that rapid drug release from nanoparticles onto the mucosal surface is beneficial for the enhancement of drug permeation, as it enables the generation of a high concentration of the drug on the mucosal surface and the achievement of a high permeation gradient across the membrane.39 However, a highly soluble drug might have limited retention time in the buccal cavity due to the salivary wash-off, thus a smaller size of nanoparticles are being targeted to be used to encapsulate drug and penetrate through the porcine sublingual epithelial membrane.40 In addition, this study demonstrated that the rate of drug permeation was steady, and similar permeability of the two final optimal formulations was observed. However, in order to optimize the permeability of the formulation, smaller particle size should be formulated, due to the fact that smaller particle size leads to larger surface area and increase the rate and amount of drug release. Additionally, permeation enhancers with higher permeability with limited toxicity could be considered to be used. For instance, permeation enhancement agents could be screened and selected, the biological compound could be conjugated to biocompatible and non-immunogenic polymers, and formulation techniques could be further investigated. Further work is recommended to support its efficacy claims by long term pharmacokinetic and pharmacodynamic studies in human beings. In particular the need for safe and effective sublingual permeation/absorption enhancers is a crucial component for a prospective future in the area of sublingual drug delivery in order to improve permeation over the sublingual mucosa, thus improving drug bioavailability.
Conclusion
The drug GSH was successfully encapsulated into PLGA nanoparticles using the double emulsion solvent evaporation method. The formulation parameters were investigated. These included different combinations of PVA and various stabilizing polymers (carbopol, MC, HPMC and poloxamer), and different concentrations of all the polymers as well as different 2nd emulsifying sonication times. Optimized formulations were generated by factorial design based on smaller particle size and higher EE as desired results. The optimized formulations were evaluated on their in-vitro drug release characteristics and ex-vivo drug permeability, and satisfactory results were obtained. Thus, we can conclude that the PLGA based double emulsion nanoparticle may provide a promising platform for the development of commercial sublingual products of GSH and the technology may be able to be applied to a wide range of protein and peptide drugs. However, in order to achieve better therapeutic effect, higher percentage of cumulative drug permeation would be expected, thereby, in future studies should include more advanced formulations involving promising permeation enhancers to be investigated.
Acknowledgements
Dr. Mary Mckendrick's contribution in proof reading the article. PLGA samples were kindly gifted by Purac Biochem company. Authors confirm that there is no conflict of interests.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Vendemiale G, Grattagliano I, Altomare E, Turturro N, Guerrieri F. Effect of acetaminophen administration on hepatic glutathione compartmentation and mitochondrial energy metabolism in the rat. Biochemical Pharmacology. 1996;52(8):1147-1154
2. Oak S, Choi BH. The effects of glutathione glycosidein acetaminophen-induced liver cell necrosis. Experimental and Molecular Pathology. 1998;65(1):15-24
3. Chi L, Ke Y, Luo C, Gozal D, Liu R. Depletion of reduced glutathione enhances motor neuron degeneration in vitro and in vivo. Neuroscience. 2007;144(3):991-1003
4. Burnside BA, Keith AD, Snipes W. Proceedings International Symposium. Control Release Bioact Mater. 1989 pp 93-94
5. Aungst BJ, Rogers NJ. Comparison of the effects of various transmucosal absorption promoters on buccal insulin delivery. International Journal of Pharmaceutics. 1989;53(3):227-235
6. Benes L, Claustrat B, Horriere F, Geoffriau M, Konsil J, Parrott KA, DeGrande G, McQuinn RL, Ayres JW. Transmucosal, oral controlled-release, and transdermal drug administration in human subjects: A crossover study with melatonin. Journal of Pharmaceutical Sciences. 1997;86(10):1115-1119
7. Brayden DJ. Controlled release technologies for drug delivery. Drug Discovery Today. 2003;8(21):976-978
8. Klose D, Siepmann F, Elkharraz K, Siepmann J. PLGA-based drug delivery systems: importance of the type of drug and device geometry. International Journal of Pharmaceutics. 2008;354(1):95-103
9. Makadia HK, Siegel SJ. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers. 2011;3(3):1377-1397
10. Peppas NA, Mongia NK. Ultrapure poly (vinyl alcohol) hydrogels with mucoadhesive drug delivery characteristics. European Journal of Pharmaceutics and Biopharmaceutics. 1997;43(1):51-58
11. Jeong B, Bae YH, Lee DS, Kim SW. Biodegradable block copolymers as injectable drug-delivery systems. Nature. 1997;388(6645):860-862
12. Vandervoort J, Ludwig A. Biocompatible stabilizers in the preparation of PLGA nanoparticles: a factorial design study. International Journal of Pharmaceutics. 2002;238(1):77-92
13. Chen G, Svirskis D, Wen J. Development and validation of a stability indicating isocratic HPLC method for gemcitabine with application to drug release from poly lactic-co-glycolic acid nanoparticles and enzymatic degradation studies. Journal of Pharmacy and Pharmacology. 2015;67(11):1528-1536
14. Costa P, Lobo JMS. Modeling and comparison of dissolution profiles. European Journal of Pharmaceutical Sciences. 2001;13(2):123-133
15. Higuchi T. Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. Journal of Pharmaceutical Sciences. 1963;52(12):1145-1149
16. Chen G, Bunt C, Wen J. Mucoadhesive polymers-based film as a carrier system for sublingual delivery of glutathione. Journal of Pharmacy and Pharmacology. 2015;67(1):26-34
17. Oeffinger BE, Wheatley MA. Development and characterization of a nano-scale contrast agent. Ultrasonics. 2004;42(1):343-347
18. Hill MW, Squier CA. The permeability of rat palatal mucosa maintained in organ culture. Journal of Anatomy. 1979;128(1):169
19. Lin H-R, Sung KC. Carbopol/pluronic phase change solutions for ophthalmic drug delivery. Journal of Controlled Release. 2000;69(3):379-388
20. Wang X, Yucel T, Lu Q, Hu X, Kaplan DL. Silk nanospheres and microspheres from silk/pva blend films for drug delivery. Biomaterials. 2010;31(6):1025-1035
21. Jose S, Juna BC, Cinu TA, Jyoti H, Aleykutty NA. Carboplatin loaded Surface modified PLGA nanoparticles: Optimization, characterization, and in vivo brain targeting studies. Colloids and Surfaces B: Biointerfaces. 2016;142:307-314
22. Kataoka K, Harada A, Nagasaki Y. Block copolymer micelles for drug delivery: design, characterization and biological significance. Advanced Drug Delivery Reviews. 2001;47(1):113-131
23. Mirhosseini H, Tan CP, Hamid NSA, Yusof S. Optimization of the contents of Arabic gum, xanthan gum and orange oil affecting turbidity, average particle size, polydispersity index and density in orange beverage emulsion. Food Hydrocolloids. 2008;22(7):1212-1223
24. Bonner JT, Savage LJ. Evidence for the formation of cell aggregates by chemotaxis in the development of the slime mold Dictyostelium discoideum. Journal of Experimental Zoology. 1947;106(1):1-26
25. Hunter RJ. A Text Book of Zeta potential in colloid science: principles and applications. Academic Press, Second Edition. 2013
26. Kirby BJ, Hasselbrink EF. Zeta potential of microfluidic substrates: 1. Theory, experimental techniques, and effects on separations. Electrophoresis. 2004;25(2):187-202
27. Takeuchi H, Matsui Y, Yamamoto H, Kawashima Y. Mucoadhesive properties of carbopol or chitosan-coated liposomes and their effectiveness in the oral administration of calcitonin to rats. Journal of Controlled Release. 2003;86(2):235-242
28. Song X, Zhao Y, Wu W, Bi Y, Cai Z, Chen Q, Li Y, Hou S. PLGA nanoparticles simultaneously loaded with vincristine sulfate and verapamil hydrochloride: systematic study of particle size and drug entrapment efficiency. International Journal of Pharmaceutics. 2008;350(1):320-329
29. Song X, Zhao Y, Hou S, Xu F, Zhao R, He J, Cai Z, Li Y, Chen Q. Dual agents loaded PLGA nanoparticles: systematic study of particle size and drug entrapment efficiency. European Journal of Pharmaceutics and Biopharmaceutics. 2008;69(2):445-453
30. Cohen-Sela E, Chorny M, Koroukhov N, Danenberg HD, Golomb G. A new double emulsion solvent diffusion technique for encapsulating hydrophilic molecules in PLGA nanoparticles. Journal of Controlled Release. 2009;133(2):90-95
31. Govender T, Stolnik S, Garnett MC, Illum L, Davis SS. PLGA nanoparticles prepared by nanoprecipitation: drug loading and release studies of a water soluble drug. Journal of Controlled Release. 1999;57(2):171-185
32. He Q, Gao Y, Zhang L, Zhang Z, Gao F, Ji X, Li Y, Shi J. A pH-responsive mesoporous silica nanoparticles-based multi-drug delivery system for overcoming multi-drug resistance. Biomaterials. 2011;32(30):7711-7720
33. Zou W, Cao G, Xi Y, Zhang N. New approach for local delivery of rapamycin by bioadhesive PLGA-carbopol nanoparticles. Drug Delivery. 2009;16(1):15-23
34. Chatzi EG, Kiparissides C. Drop size distributions in high holdup fraction dispersion systems: effect of the degree of hydrolysis of PVA stabilizer. Chemical Engineering Science. 1994;49(24):5039-5052
35. Faisant N, Siepmann J, Benoit JP. PLGA-based microparticles: elucidation of mechanisms and a new, simple mathematical model quantifying drug release. European Journal of Pharmaceutical Sciences. 2002;15(4):355-366
36. Klose D, Siepmann F, Elkharraz K, Krenzlin S, Siepmann J. How porosity and size affect the drug release mechanisms from PLGA-based microparticles. International Journal of Pharmaceutics. 2006;314(2):198-206
37. Ritger PL, Peppas NA. A simple equation for description of solute release I. Fickian and non-Fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. Journal of Controlled Release. 1987;5(1):23-36
38. Korsmeyer RW, Gurny R, Doelker E, Buri P, Peppas NA. Mechanisms of solute release from porous hydrophilic polymers. International Journal of Pharmaceutics. 1983;15(1):25-35
39. Mudgil M, Pawar PK. Preparation and in vitro/ex vivo evaluation of moxifloxacin-loaded PLGA nanosuspensions for ophthalmic application. Scientia Pharmaceutica. 2013;81(2):591
40. Chen G, Svirskis D, Lu W, Ying M, Huang Y, Wen J. N-trimethyl chitosan nanoparticles and CSKSSDYQC peptide: N-trimethyl chitosan conjugates enhance the oral bioavailability of gemcitabine to treat breast cancer. Journal of Controlled Release. 2018May10;277:142-53
Author contact
![]() Corresponding author: J. Wen. Address: School of Pharmacy, University of Auckland. 85 Park Road, Grafton, Auckland, New Zealand. E-mail address: j.wenac.nz (J. Wen)
Corresponding author: J. Wen. Address: School of Pharmacy, University of Auckland. 85 Park Road, Grafton, Auckland, New Zealand. E-mail address: j.wenac.nz (J. Wen)