J Biomed 2018; 3:68-73. doi:10.7150/jbm.27245 This volume Cite
Review
New insight into the genetics, developmental mechanism and therapeutic targets of Retinitis Pigmentosa
Luqman Khan1, ![]() , Qaisar Hanif2, Muhammad Usman3
, Qaisar Hanif2, Muhammad Usman3
1. Graduate School of Life Sciences, Tohoku University, Sendai, Japan
2. Department of Ophthalmology Allied Hospital, Medical University, Faisalabad, Pakistan
3. Department of Bioinformatics & Biotechnology, Government College University Faisalabad, Pakistan
Received 2018-5-14; Accepted 2018-8-16; Published 2018-9-4
Abstract
Hereditary degeneration of the human retina is genetically miscellaneous, with more than a hundred genes identified until now. Retinitis pigmentosa (RP) is a group of genetic retinal disorder which results in the degeneration of rods & cones photo-receptor. Retinitis pigmentosa is categorized by moon blindness and gradual loss of peripheral visualization, many time leads to thorough blindness. This disorder is both genetically and clinically heterogeneous. Retinitis pigmentosa could be linked with the syndromic disease as genes important for the retina also play the role in complementary tissues. Improvements in molecular genetics have delivered novel insights into the genes responsible and the pathogenic tools of retinitis pigmentosa. The inheritances of retinitis pigmentosa are multidimensional, and the disorder segregates following different kinds of transmissions. Treatments like gene therapy & stem cell treatment possibly will provide possible treatment probability for patients with retinitis pigmentosa.
Keywords: Retinitis pigmentosa, Genetic architecture, Retina, blindness
Introduction
Eyesight is the common sense that we give importance extra than that of the other senses. Our eyes are called windows to the soul. The eye is really magnificent and most complex organ of the human body. Only one-sixth of the eye exterior is visible. Besides contributing a focal point to straight one's consideration when talking to another person, eyes brings in further 90 % of the information in going to the average human brain. Congenital eye diseases contain 1/3 of all reported human genetic disorders [1].
Retinitis Pigmentosa is a far-reaching class of disorders disturbing the retina [2]. For above 140 years, this disorder has been the tag given to a group of congenital progressive disorders of the retina by means of a mutual prevalence of just about 1/5000 [3]. Retinitis Pigmentosa is an extremely diverse class of disorders which is categorized by the deposits of the retinal pigment in the middle edge of the retina. An individual with a retinal impairment is commonly analyzed with retinitis pigmentosa based on nightly sightlessness which happens early stages of lifespan following channel vision late in a lifetime [4]. Retinitis Pigmentosa is transferred to offspring in a diverse fashion including autosomal dominant, autosomal recessive, X-linked, digenic and mitochondrial [5].
Still no cure is available for RP. Most important hindrances for the efficacious modified therapy improvement are the inadequate considerate of cellular malady mechanisms and the gigantic heterogeneity of ailment-causing transmutations (Figure 1).
Clinical Assessment and Finding
Retinitis Pigmentosa is exceedingly a mutable condition. Round about all patients developed symptomatic optical damage in early stages of life whereas some remain asymptomatic till the teenage of life. Lots of patients fall into the archetypal mode of complexity with black adaptation and nocturnal loss of sight in adolescence and mid-peripheral optical field in early stages of life. With the progress in the disorders they loss peripheral vision, finally, established a tunnel visualization and eventually loss central vision generally at the age of 60. The common sign of retinitis pigmentosa night-time loss of sight, soreness of the retina, the obvious limitation of the field of visualization etc. [6]. Symptoms depend on whether rod or cones are initially involved. Most forms of RP initially result in degeneration of rod cells, which sometimes called rod-cone dystrophy mostly initiates with night blindness.
As the disease progress more and more cells of rod degenerate, the patient lost their peripheral vision. An individual with retinitis pigmentosa mostly experienced a loop of vision loss in their mid-periphery with small Landmass of loss in their very far periphery. Other patients report the sensation of channel vision, as though they see the globe through a hay. Numerous individual with RP maintains a small amount of central vision throughout the entire lifespan (Figure 2).
Genetics of the Retinitis Pigmentosa
The 20th century experienced numerous inventions and developments in medical science than all previous decades as a whole. The fundamental event was the genetics revolution, starting with discovery of DNA structure in 1953 followed in 1980 by the capability to chemically read the genetic code, isolation of particular gene and clone for more studies. Over the past decade, more than 40 genes resulting retinal impairments in Human beings have been mapped, typically by linkage studies [8].
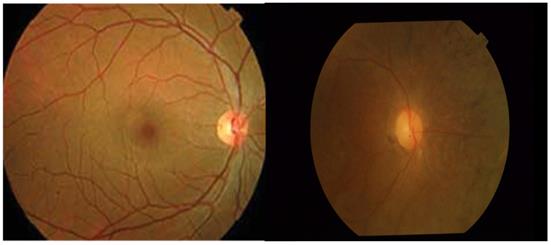
Typical image of Fundus of healthy and RP affected retina (From left to Right).
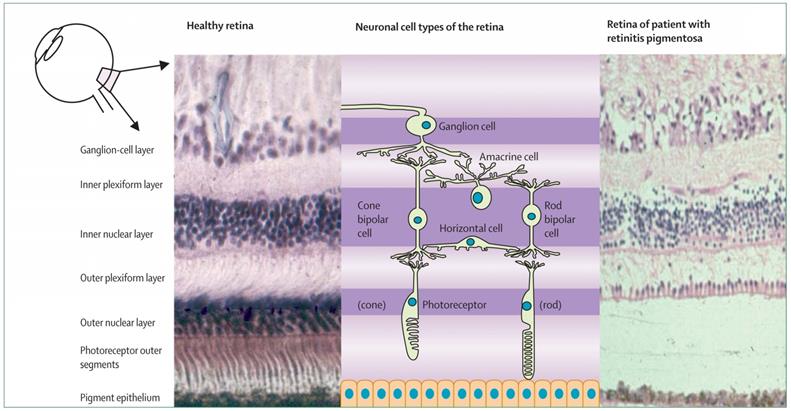
Histological presence of normal human retina (left) and retina of an individual with retinitis pigmentosa at a mid-stage of disease (right) [7].
The genetics of RP is complicated, Retinitis Pigmentosa can be sub-classified into various genetic categories. Most of RP is monogenic but the digenic mode of inheritance is also known [9]. Percentage of hereditary manner revealed that all isolated cases such as the patient with not any other affected relative are autosomal recessive, though some can show novel dominant mutation, illustrations of uniparental isodisomy [7] or for male X-linked mutation. Non-Mendelian hereditary manner such as digenic in addition to parental inheritance have been investigated, but possibly result in only a minor percentage of cases. Thus the pattern of inheritance cannot be determined for all patients but it is assumed that all cases have the genetic basis [10].
Autosomal dominant Retinitis Pigmentosa
This form is generally the slightest type of Retinitis Pigmentosa, using several of the cases not starting up until the late periods of the lifecycle. Off total, 15 to 20 percent of the total cases of retinitis pigmentosa remain autosomal dominant [9]. This type of retinitis pigmentosa is resulted in the minimum of 12 genes, containing PRPF31 and RP [11]. 14 loci have been recorded for autosomal dominant RP, with many genes to be copied. The initial gene duplicated for retinitis pigmentosa was Rhodopsin (RHO) recognized in 1990 [12]. Aforementioned gene is too the chief causal gene for autosomal dominant Retinitis Pigmentosa.
Autosomal Recessive Retinitis Pigmentosa
In distinction to autosomal dominant RP, the autosomal recessive retinitis pigmentosa is characteristically stated in teenage individuals and extremity is significantly higher. 25 linked loci for autosomal recessive RP loci are reported & for 22 the causal alteration has been recognized. These changes are mostly liable for a minor proportion of cases of around 1 to 5 % [13]. Autosomal recessive RP is one of the most wide-ranging sorts of Retinitis pigmentosa throughout the world and results for around 39% of the total cases in Spanish people [14]. A number of the autosomal recessive RP genes are too associated with other diseases of the retina. One of the causative gene for such disorder is CRB1; changes in this particular gene is to result in Leber congenital amaurosis (LCA), which is a new clinically diverse ailment of the retina [15].
X-linked Retinitis Pigmentosa
X- linked chromosome associated Retinitis Pigmentosa that all the time commenced with heavy signs, primary onset and swift worsening, the result for 6 to 17% of hereditary Retinitis Pigmentosa cases [16]. Till now 5 inbred loci have been planned for X-linked Retinitis Pigmentosa, comprising RPGR, RP2, RP6, RP23 and RP26. Alterations in the RPGR gene are anticipated to result in 15 to 20 percent of total circumstances of RP, larger than any superfluous particular RP locus [17].
Digenic Retinitis Pigmentosa
Digenic inheritance is the associations of transformations of two contradictory genes causing in a communicable eye malady. In such condition alterations in Retinal Outer Segment Membrane Protein 1(ROM1) and RDS gene united to result in a digenic hereditary phenotype [18].
Identified Gene for the Retinitis Pigmentosa
The RP25 Gene
The RP25 gene is the twenty sixth gene to be recognized for an autosomal recessive retinitis pigmentosa [19]. The locus for RP25 was previously identified through homozygosity mapping by leading functional candidate genes. Four Spanish pedigree with arRP were mainly connected to an area on chromosome 6q stuck between microsatellite markers D6S257 and D6S1644 [20]. The homozygosity mapping in 145 patients with autosomal recessive retinitis pigmentosa is recognized with a pooled division of homozygosity on chromosome 6 in two affected sibs that corresponded with the RP25 locus. Six anticipated genes contained by the RP25 interim on chromosome 6q12 in ten Spanish retinitis pigmentosa effected families that were previously linked to the RP25 interval and recognized homozygosity or miscellaneous heterozygosity for six poles apart mutations in the gene they designated EYS [21].
Mapping of the RP25 Gene
A novel locus for autosomal recessive retinitis pigmentosa (arRP), located on 6q between markers D6S257 and D6S1644, in an area that contains genes coding subunits GABRR1 (137161) and GABRR2 (137162) of the GABA-C receptor, which is the effector of lateral inhibition in the retina [22]. Without a doubt, the study was embarked on a candidate gene basis; the candidate designated chromosomal area including genes that code the transformed subunits of the GABA receptors for homozygosity mapping in hereditary peoples affected by autosomal recessive retinitis pigmentosa. The GABRR1 and GABRR2 genes are to be found at 6q14-q21 [23].
The RPE65 Gene
The membrane connected form of RPE65 (mRPE65) is triply palmitoylated and is a chaperone for all-trans-retinyl esters, allowing their entrance into the visual cycle for transforming into 11-cis-retinal[24]. The soluble form of RPE65 (sRPE65) is not palmitoylated and is a chaperone for vitamin A as a substitute of all-trans-retinyl esters. As a consequence, the palmitoylation of RPE65 controls its ligand binding selectiveness. The two chaperones are changed with lecithin retinol acyltransferase (LRAT; 604863) functioning as a molecular switch, with mRPE65 as the palmitoyl donor. When the chromophore production is not obligatory, mRPE65 is changed into sRPE65 by LRAT, and furthermore, the chromophore production is obstructed. The studies revealed an inventive character for palmitoylated proteins as molecular switches and for LRAT as a palmitoyl transferase whose role is to catalyze the alteration of mRPE65 to sRPE65[25].
In the internal of the visual cycle, an isomer hydrolase is responsible for isomerization and hydrolysis of the all-trans-retinyl ester to 11-cis retinol, and LRAT be responsible for the retinyl ester substrate. Human recombinant RPE65, when co-expressed with LRAT in human embryonic kidney cells or COS-1 cells, efficiently produced 11-cis retinol from the all-trans-retinyl ester [26]. Enzymatic activities were linearly dependent on the expression level of RPE65.
Mapping of the RPE65 Gene
Using a human/hamster hybrid panel mapped the human RPE65 gene to chromosome 1 as well as, by fluorescence in situ hybridization, refined the localization to chromosome 1p31[27]. Through the research investigation of rodent/human somatic cell hybrids and by fluorescence in situ hybridization, confirmed the assignment to chromosome 1p31[28]. Through the interspecific backcross examination, mapped the mouse Rpe65 gene to the distal portion of chromosome 3[29].
Diagnosis and Therapeutics
The warning sign of Retinitis Pigmentosa may bear a resemblance to other maladies. So far for the identification of Retinitis Pigmentosa, comprehensive ophthalmological consideration might be exceedingly indicative. This is without doubt, precise exclusively in initial times of life. The subsequent test is implemented on the affected persons.
Fundus Finding
Ophthalmoscopy of the retina with cutting-edge Retinitis Pigmentosa is categorized by the manifestation of the intraretinal cluster of obscure pigment, prominently alleviated retinal vessel, damage of retinal pigment epithelium and pallor of the ophthalmic nerve. This variation repels a long time retinal deterioration and not be existent to form the diagnosis of the Retinitis Pigmentosa. The fundus finding, although are instrumental in differentiating Retinitis Pigmentosa from other retinal conditions that have the identical clinical result but differentiating retinal variation.
Visual Acuity Test
Visual fields, dignified with a Goldmann perimeter or a Humphrey field analyzer (Carl Zeiss, Dublin, CA, USA), characteristically have scotoma in the middle-periphery which amplify over years attributable to damage of rods and cones objectives. On average to progressive RP, only minor landmasses of visualization keep on in the faraway marginal field and in the optical axis, later these regions of vision gradually vanish.
Color Vision
This examination was achieved by typical Ishihara plates. Color vision evaluated with the help of Ishihara plates. Other investigations might display standard color vision or an insufficiency of blue cone function. It is essential to consent that these prospective methods are not conjointly exclusory and that few of them are probable to act in concerting the cell death of photoreceptor. Moreover, diverse methods are unlikely to be intricate for dissimilar classes of alteration.
With the understanding of causative genes in more than half of affected individuals with Retinitis pigmentosa and accumulative understanding about accompanying biochemical shortcomings, numerous clinicians are positive that innovative management for the disorder will quickly be developed. Several systematically miscellaneous tactics to treat RP are being explored. Some of the modern treatments are discussed below.
Stem Cell Transplantation
Stem cell therapy has been deliberated a favourable approach in the management of retinal disorders. Human embryonic stem cells (hESCs) were formerly measured the merely auspicious basis for substitution of the cells in reformative medication. Nevertheless, Human embryonic stem cells are linked with various disadvantages, comprising the simultaneous organization of lifetime immunosuppressive analysis and narrow usefulness. Hence, patient specified induced pluripotent stem cell (iPSC) therapy was developed, assessment of ailment pathophysiology, innovative drug improvement, and the probability of stem cell treatment in retinal sicknesses was incessantly improved.
The retina, predominantly the subretinal cosmos, is valuable to stem cell transplantation because the eye is comparatively immune restricted [30]. The blood visual obstacle guards the subretinal universe by antigen-specific inhibition of reactions of cellular and humoral immune coordination, providing that it is not bodily cooperated in transplantation or due to the core disease pathology. In such circumstances, the immunogenicity leftovers a challenge in Human embryonic stem cells derived transplantation, however, can be allayed by the induced pluripotent stem cell method. The predilection for tumorigenesis; such that the development of teratomas, as is repeatedly the situation with induced pluripotent stem cell, which is susceptible to epigenetic and transcriptional abnormalities[31]. Moreover, the eye is effortlessly manageable for observing by assessment and with more than a few high-resolution imaging modalities, deprived of the requirement for tissue removal pre and post-transplantation.
Gene Therapy
Ten percent of the human congenital disorders are associated with a hereditary retinal dysfunction. Gene therapy for this purpose will almost certainly be a future crucial therapeutic preference [32]. Gene therapy is a technique that replaced or turns off the mutated disorder producing gene to repair some typical protein persistence. In the congenital diseases, like retinitis pigmentosa, a lot of measures are used to substitute or treat abnormal genes: (a) insertion of a characteristic gene into the genome to exchange non-functional or un-healthy genes by means of a carrier vector, (b) ribozyme therapy, and (c) RNA interference. Gene regeneration is critical in recessive conditions, nevertheless, ribozyme therapy and RNA interference may be valuable in autosomal dominant conditions. In the dominant conditions, even and uneven gene produces (proteins) are formed by the regular and transformed gene consistently. The abnormal gene product is unfavourable to the photoreceptors and ultimately fallouts in cell damage [33]. Ribozymes could be aimed to divide mutated mRNA molecules so that the damaging protein could not be manufactured, in that way protecting the cells. Even though ribozymes may not eradicate all transmuted mRNA, this failure was revealed to be suitable for the protection of vision in an autosomal dominant RP canine model. RNA interference working in a parallel way, causing destruction of the abnormal RNA by existing cell defence manners.
Advances in gene therapy treatment have been emerging in clarifying visual function. For autosomal recessive RP a counteractive gene may be transported together into the cell over the use of a recombinant adenovirus (or adeno-associated virus) vector, in hopes the virus may deliver the normal gene to the host's cell, replacing the diseased gene. These techniques have been tentatively demonstrated to postponed and even reverse the development of retinitis pigmentosa with complementary improvement of photoreceptor function in several animal simulations [34].
Conclusion
Retinitis pigmentosa (RP) refers to a set of congenital disorders in which abnormalities of the photoreceptors of the retina causing to progressive optical loss. Advances in the pathophysiology of retinitis pigmentosa are producing novel diagnoses for the treatment of retinitis pigmentosa. Future treatments for retinitis pigmentosa are considerably anticipated, particularly for genetically well-defined subsets of patients, because of newly recognized genes, increasing awareness of affected biochemical passageways, and development of animal model & treatments like gene therapy & stem cell treatment possibly will deliver potential treatment chances for patients with retinitis pigmentosa.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Khan L, Ali M, Qasim M, Jabeen F, & Hussain B. Molecular basis of glaucoma and its therapeutical analysis in Pakistan: an overview. BMRAT. 2017;4(03):1210-1227
2. Carmichael K.D. Play Therapy With Children With Disabilities. Handbook of Play Therapy. 2015:397
3. Zrenner E. Retinitis Pigmentosa. In Encyclopedic Reference of Genomics and Proteomics in Molecular Medicine. Springer. 2005:1643-1648
4. Sardegna J. The encyclopedia of blindness and vision impairment. Infobase Publishing. 2002
5. Annunen-Rasila J. Molecular and Cell Phenotype Changes in Mitochondrial Diseases. 2007: University of Oulu.
6. Jones B. et al. Retinal remodeling in human retinitis pigmentosa. Exp Eye Res. 2016;150:149-165
7. Hartong D.T. et al. Retinitis pigmentosa. The Lancet. 2006;368(9549):1795-1809
8. Neveling K. et al. Identification and analysis of inherited retinal disease genes. Retinal Degeneration: Methods and Protocols. 2013:3-23
9. Rivolta C. et al. Retinitis pigmentosa and allied diseases: numerous diseases, genes, and inheritance patterns. Hum Mol Genet. 2002;11(10):1219-1227
10. Daiger SL. Sullivan, and S. Bowne, Genes and mutations causing retinitis pigmentosa. Clin. Genet. 2013;84(2):132-141
11. Bujakowska K. et al. Study of gene-targeted mouse models of splicing factor gene Prpf31 implicated in human autosomal dominant retinitis pigmentosa (RP). Invest Ophthalmol Vis Sci. 2009;50(12):5927-5933
12. Tam B.M, Moritz O.L. Dark rearing rescues P23H rhodopsin-induced retinal degeneration in a transgenic Xenopus laevis model of retinitis pigmentosa: a chromophore-dependent mechanism characterized by production of N-terminally truncated mutant rhodopsin. J Neurosci. 2007;27(34):9043-9053
13. Audo I. et al. EYS is a major gene for rod-cone dystrophies in France. Hum mutat. 2010:31 (5)
14. Sherwin J.C. et al. Genetic isolates in ophthalmic diseases. Ophthalmic Genet. 2008;29(4):149-161
15. Cwerman-Thibault H. et al. Gene therapy for mitochondrial diseases: Leber Hereditary Optic Neuropathy as the first candidate for a clinical trial. C R Biol. 2014;337(3):93-206
16. Goodwin P. Hereditary retinal disease. Curr Opin Ophthalmol. 2008;19(3):255-262
17. Bader I. et al. X-linked retinitis pigmentosa: RPGR mutations in most families with definite X linkage and clustering of mutations in a short sequence stretch of exon ORF15. Invest Ophthalmol Vis Sci. 2003;44(4):1458-1463
18. Nguyen O.N.P. Molecular characterization of the interaction between peripherin-2 and opsins in rod and cone photoreceptors. 2016;lmu.
19. Bonilha V.L. et al. Histopathological comparison of eyes from patients with autosomal recessive retinitis pigmentosa caused by novel EYS mutations. Graefes Arch Clin Exp Ophthalmol. 2015;253(2):295-305
20. O'Driscoll C. Autosomal recessive retinitis pigmentosa, identification and partial characterisation of a novel gene implicated in RP25. 2010;UCL (University College London).
21. Barragán I. et al. Mutation spectrum of EYS in Spanish patients with autosomal recessive retinitis pigmentosa. Hum mutat. 2010:31 (11)
22. Ruiz A. et al. A Major Locus for Autosomal Recessive Retinitis Pigmentosa on 6q, Determined by Homozygosity Mapping of Chromosomal Regions That Contain Gamma-Aminobutyric Acid-Receptor Clusters. Am J Hum Genet. 1998;62(6):1452-1459
23. Marcos I. et al. Mutation analysis of GABRR1 and GABRR2 in autosomal recessive retinitis pigmentosa (RP25). J Med Genet. 2000;37(6):e5-e5
24. Xue L. et al. A palmitoylation switch mechanism in the regulation of the visual cycle. Cell. 2004;117(6):761-771
25. Maiti P. et al. Small molecule RPE65 antagonists limit the visual cycle and prevent lipofuscin formation. Biochemistry. 2006;45(3):852-860
26. Moiseyev G. et al. RPE65 is the isomerohydrolase in the retinoid visual cycle. Proc Natl Acad Sci U S A. 2005;102(35):12413-12418
27. Weith A, Vance J. Report of the second international workshop on human chromosome 1 mapping 1995. Cytogenet Genome Res. 1996;72(2-3):113-154
28. Kabir F. et al. Novel mutations in RPE65 identified in consanguineous Pakistani families with retinal dystrophy. Mol vis. 2013;19:1554
29. Hamel C.P. et al. The gene for the retinal pigment epithelium-specific protein RPE65 is localized to human 1p31 and mouse 3. Geno. 1994;20(3):509-512
30. Mountford J. Human embryonic stem cells: origins, characteristics and potential for regenerative therapy. Transfus Med. 2008;18(1):1-12
31. Rodrigues M. Life and death of mesenchymal stem cells/multipotential stromal cells (MSC): Countervailing regulation by survival and apoptotic signaling. University of Pittsburgh. 2012
32. Balaggan K, Ali R. Ocular gene delivery using lentiviral vectors. Gen the. 2012;19(2):145-153
33. Van Hooser J.P. et al. Rapid restoration of visual pigment and function with oral retinoid in a mouse model of childhood blindness. Proc Natl Acad Sci U S A. 2000;97(15):8623-8628
34. Beckerman M. Modeling and Simulation of Microelectrode-Retina Interactions. Oak Ridge Y-12 Plant, TN (US). 2002
Author contact
![]() Corresponding author: Luqman Khan. e-mail: luqman.zoologycom
Corresponding author: Luqman Khan. e-mail: luqman.zoologycom